Research

Research Overview
Work in the ScleroLab is patient-oriented, bench-to-bedside, and encompasses several approaches.
Work in the ScleroLab is patient-oriented, bench-to-bedside, and encompasses several approaches, ranging from genetics and functional genomics to vascular biology, cellular immunology, metabolic studies, molecular and live imaging, and matrix biology. We deploy these approaches to discover, validate, and develop novel targets for diagnosis, precision medicine, and targeted human therapies. Our current research projects are listed below.
The overarching theme of our future work is to understand how gene-environment interactions trigger and sustain fibrosis, and to mine this knowledge for drug discovery. We will pursue four linked areas:
- (dys)regulation of NAD+ metabolism linking inflammation, vascular injury and fibrosis in scleroderma
- genetic polymorphisms such as A20 (TNFAIP3) shaping immunity and matrix remodeling in health and disease
- diet and gut microbiome-derived metabolites, such as trimethylamine oxide (TMAO), driving systemic immune, vascular and fibrotic responses
- a novel pathogenic acquired primary ciliopathy in scleroderma
Our work in each of these areas is cross-disciplinary, patient-oriented, and therapy-focused. We will make extensive use of human disease samples and clinical information collected from our rheumatology clinics at Michigan Medicine. We will identify and functionally characterize endophenotype-associated common and rare genetic variants; study gene expression in biopsies using bulk and single-cell RNA-seq and ATAC-seq, spatial transcriptomics and metabolomics; and optimize disease models including cellular assays, organoids and engineered mice. We will collaborate with investigators from several departments using cross-cutting approaches of computational biology, cell biology, immunology, and bioengineering.
Scleroderma, along with lupus and other chronic rheumatic diseases, is characterized by synchronous injury and damage in the immune, vascular and stromal compartments throughout the body. Understanding and treating these conditions, therefore, requires integrated “organ-agnostic” cross-cutting scientific collaborations.
(Sponsor: NIH; R01 AR074997)
This project focuses on the role of sterile inflammation, fibroblast activation, and persistent myofibroblasts accumulation in progressing organ fibrosis in systemic sclerosis/scleroderma (SSc). Tissue damage from chronic injury causes local generation and accumulation of endogenous TLR4 ligands called “damage-associated molecular patterns (DAMPs)”, such as fibronectin-EDA and tenascin-C.
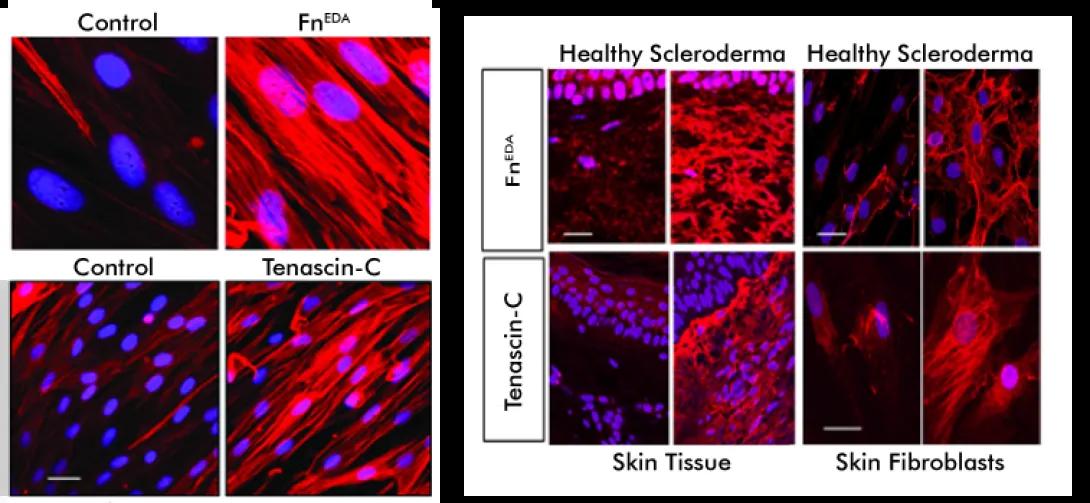
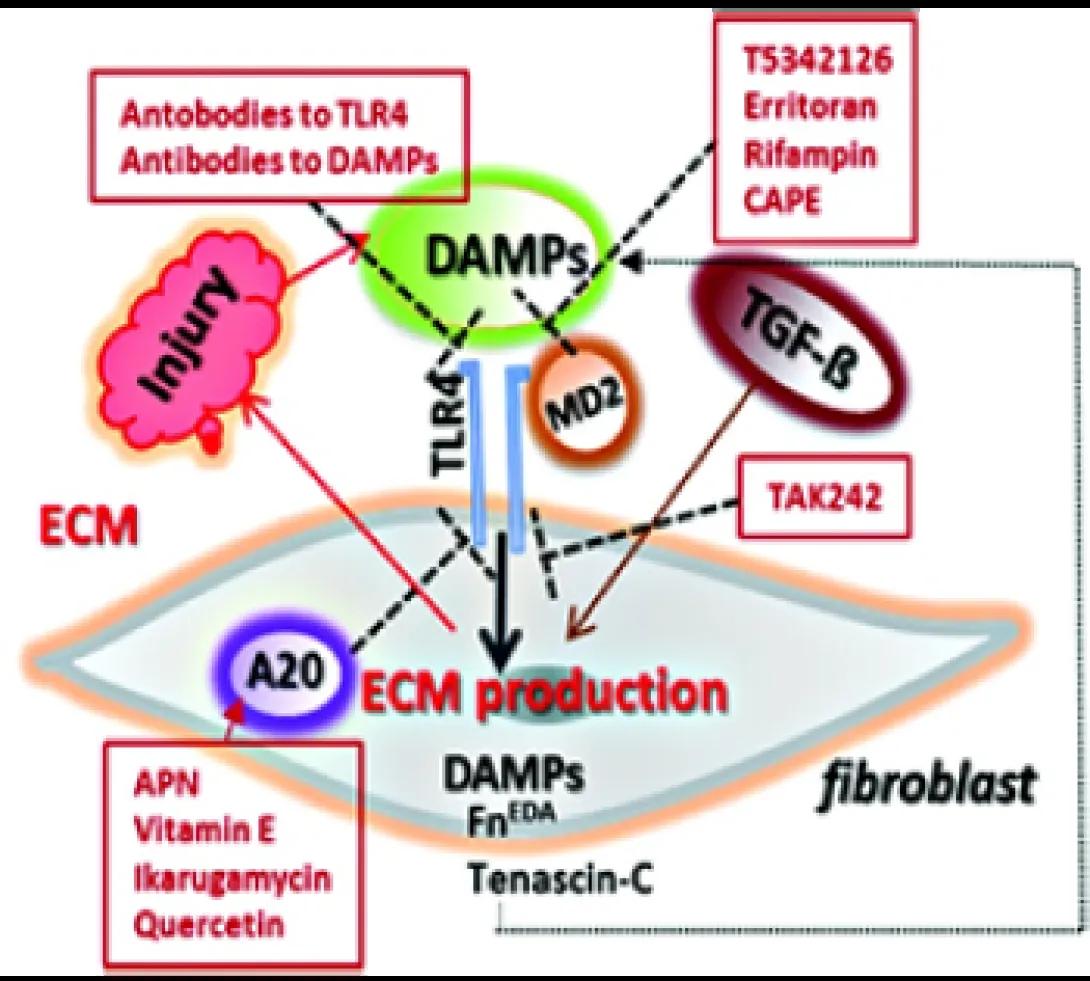
DAMPs, in turn, activate innate immune signaling in resident fibroblasts through TLR4. This results in enhanced matrix production and TGF-beta secretion, establishing a self-amplifying vicious cycle of fibrosis.
We are interested in the underlying mechanisms of self-sustaining fibroblast activation. We study fibrosis in various mouse models which may be translated into therapeutic approaches for treating fibrotic disease, and delineate predictive biomarkers of ongoing innate immune signaling that can enable a precision medicine approach to treating SSc, a disease that exhibits marked clinical and molecular heterogeneity.
We are also interested in the role of cell-intrinsic protective mechanisms that normally act as the brakes on fibroblast activation, such as A20/TNFAIP3, which are hypofunctional in SSc, further aggravating unopposed TLR4 signaling and fibrogenesis.
(Prime Sponsor: NIH-NIAMS. Direct Sponsor: TeneoBio, Inc.)
Age-related pathologies are associated with organismal decline in nicotinamide adenine dinucleotide (NAD+) that is due to dysregulation of NAD+ homeostasis and involves the NADase CD38.
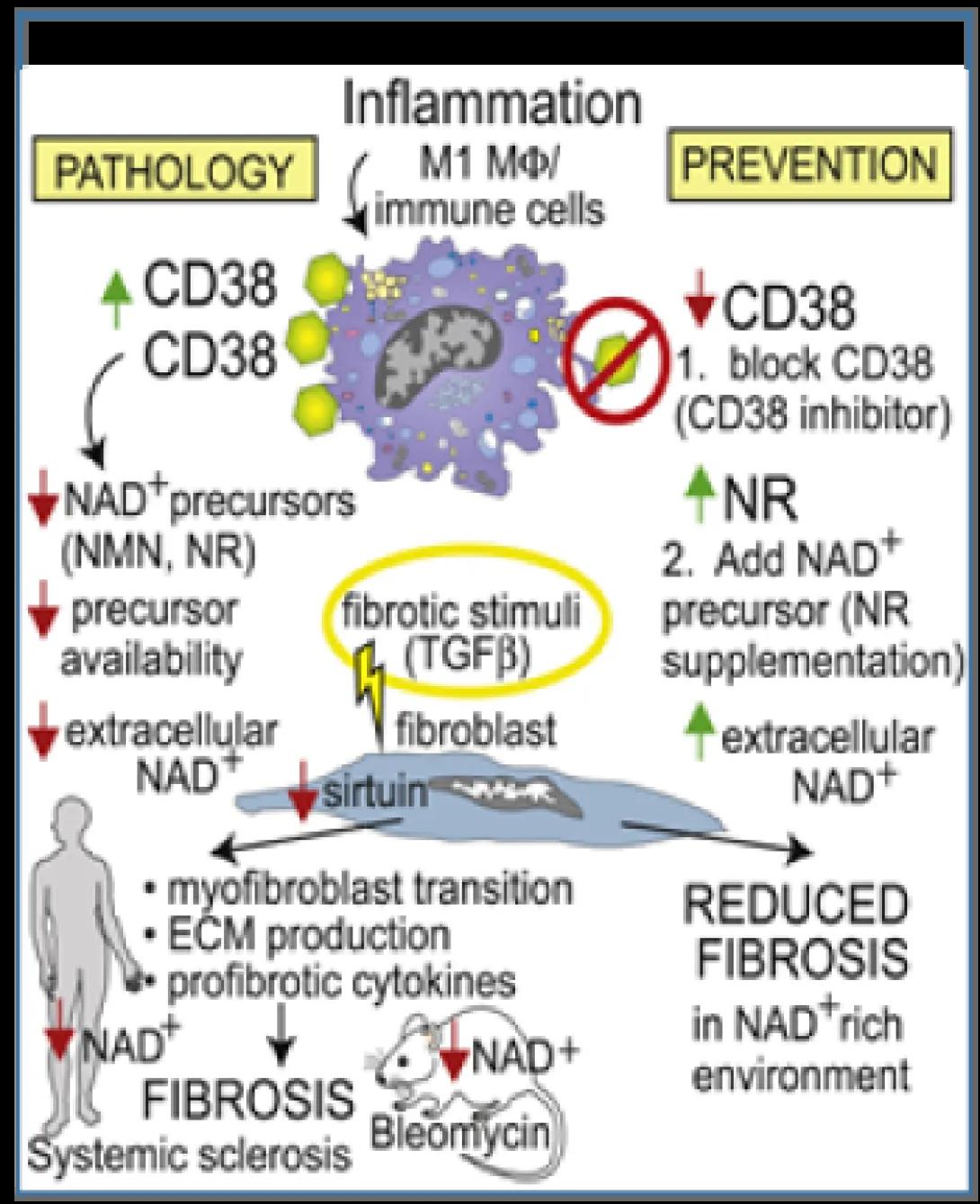
We now show that CD38 is upregulated in patients with diffuse cutaneous SSc, and CD38 levels in the skin associate with molecular fibrosis signatures, as well as clinical fibrosis scores, while expression of key NAD+ synthesizing enzymes is unaltered. Boosting NAD+ via genetic or pharmacological CD38 targeting or NAD+ precursor supplementation protected mice from multiple organ fibrosis. CD38 was found to reduce NAD+ levels and sirtuin activity to augment cellular fibrotic responses, while inhibiting CD38 had the opposite effect. Thus, CD38 upregulation and resulting disrupted NAD+ homeostasis as a fundamental mechanism driving fibrosis in SSc, suggesting that CD38 might represent a novel therapeutic target.
- TREM-1 Inhibitor for the Treatment of Scleroderma. Prime Sponsor: NIH-NIAMS. SBIR. Direct Sponsor: SignaBlok, Inc.
- Idiopathic Pulmonary Fibrosis-Associated SNPs in Scleroderma Interstitial Lung Disease. Sponsor: Boehringer Ingelheim/Columbia University
- Metaorganismal TMAO Pathway Driving Scleroderma Pathogenesis: Novel Gene-Environment Interaction Paradigm and Therapeutic Target. Sponsor: NIH R61 AR076821
- Dysregulated Ciliogenesis in Scleroderma: Novel Pathogenic Mechanism. Sponsor: Rheumatology Research Foundation
- Altered SPAG17 Signaling in SSc: A Novel Mechanism for SSc-lLD and a Therapeutic Target. Prime Sponsor: Boehringer Ingelheim. Direct Sponsor: Virginia Commonwealth University (VCU). U-M is subrecipient of subK from VCU
- IL-23 Signaling and Its Modulation in Scleroderma (Systemic Sclerosis): Novel Therapy. Sponsor: Sun Pharma Global FZE
- Scleroderma Renal Crisis as a Genetic Complementopathy. Prime Sponsor: NIH-NIAMS. R21. Direct Sponsor: Washington University at St. Louis (WUSTL). U-M is subrecipient of subK from WUSTL
- Idiopathic Pulmonary Fibrosis-Associated SNPs in Scleroderma Interstitial Lung Disease. Prime Sponsor: Boehringer Ingelheim (Bernstein, Varga). Direct Sponsor: Columbia University (Elana Bernstein). U-M is subrecipient of subK from Columbia University
- Comprehensive Characterization and Contribution of the Keratinocytes-Fibroblasts Crosstalk in 3D-Scleroderma Skin Equivalent. Sponsor: Boehringer Ingelheim