Research | Barnett Lab

Barnett Lab Overview
Investigating the molecular cues that govern viral disease progression
Viral diseases, such as those caused by severe acute respiratory coronavirus 2 (SARS-CoV-2), vary widely between individuals. While some people infected with SARS-CoV-2 never develop symptoms, others face life-threatening illness or long-term chronic disease. In the Barnett lab, we seek to understand how these disparate disease states emerge during viral infection and develop therapeutic interventions.
A major factor guiding disease progression is the innate immune response, which may promote clearance of viral pathogens or exacerbate disease depending on the timing and nature of the response. The innate immune response relies on proteins known as pattern recognition receptors (PRRs) to recognize viral infection and activate inflammatory and antiviral signaling pathways. PRRs detect two kinds of molecules during infection: pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). PAMPs are molecules derived from the pathogen, such as viral genomic RNA or viral proteins, whereas DAMPs are molecules derived from the host as a consequence of pathogen activity, such as DNA released from the mitochondria or nucleus. While viral PAMPs alone stimulate innate immune responses, our work has shown that viral DAMPs amplify and alter the innate immune response during infection, increasing the inflammatory response through the activation of additional signaling pathways. These findings underscore the importance of host-derived responses in viral infection, mandating further interrogation of the role of DAMPs in viral disease states.
One potential source of DAMPs during viral infection is cell death, and SARS-CoV-2 and other RNA viruses stimulate cell death in human hosts during infection. Our research found that cell death response differs between SARS-CoV-2 variants-of-concern (VOC) and correlates with disease progression. In particular, we found that the Delta VOC (B.1.617.2), which caused more severe disease in humans and in mouse models, induced higher levels of cell death at faster rates than the Omicron BA.1 VOC (B.1.1.529), which caused milder disease in humans and murine models. This suggests that the cell death response to viral infection influences pathogenesis, making it essential to understand this host-derived response during infection and its relationship with DAMP production and the immune response.
With this perspective on host-pathogen interactions, our group investigates how viruses cause cell death and damage and how virus-induced cell death and damage informs the immune response and disease progression with an emphasis on SARS-CoV-2 infection. By understanding these processes, we can pinpoint key molecular interactions for therapeutic targeting to prevent and treat harmful viral disease states. We are investigating these topics using primary human organotypic culture systems, murine models, cell lines, and patient samples to ensure our work extends throughout all levels of biology (molecular, cellular, and organismal) and is relevant to human health. As our research progresses, we aim to identify molecular interactions specific to high-risk patient groups and those shared between viruses to create broad-spectrum therapeutics for existing and emerging pathogens.
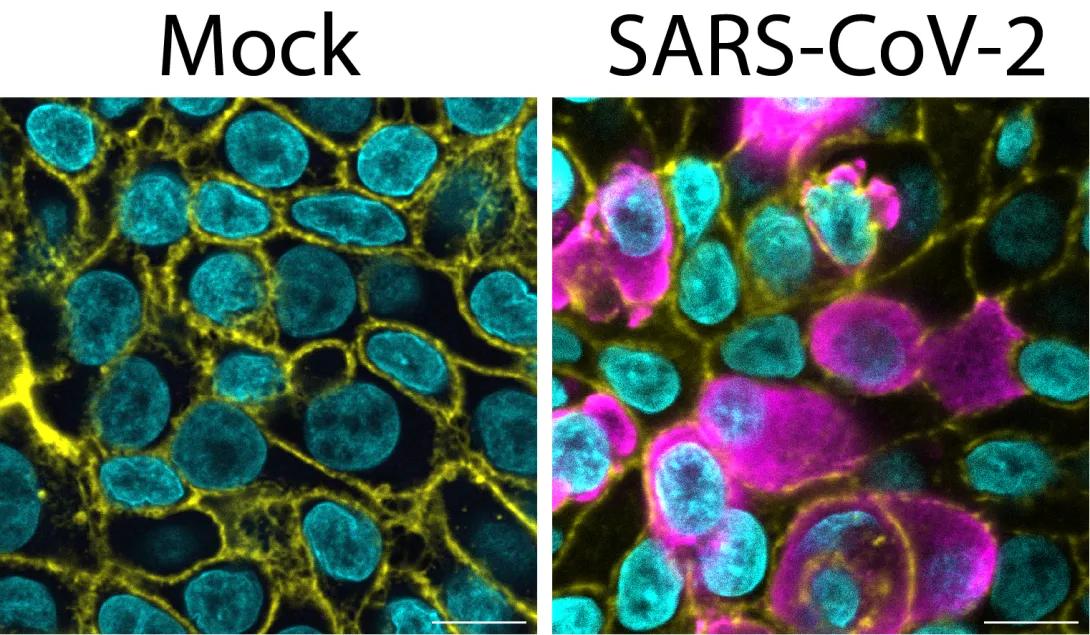
These are images of primary human airway epithelia without SARS-CoV-2 infection on the left and with SARS-CoV-2 infection on the right. SARS-CoV-2 nucleocapsid is shown in magenta, nuclei are shown in cyan, and filamentous actin, which shows the cell periphery, is shown in yellow.
PROJECTS IN THE BARNETT LAB
Exaggerated inflammatory cytokine responses, including interleukin-1β (IL-1β) and IL-6, are linked to acute respiratory distress syndrome (ARDS), a form of lung failure observed in severe COVID-19. Our recent work showed that crosstalk between the SARS-CoV-2-infected airway epithelium and myeloid cells promotes inflammasome activation and IL-1β release, which in turn stimulated IL-6 secretion (Barnett et al., Cell Host & Microbe, 2023). This response was driven by genomic and mitochondrial DNA DAMP release from infected airway epithelial cells, because these DNA DAMPs were then detected by immune cells to stimulate inflammasome activation and IL-1β release. In turn, this response led to IL-6 release. This showed that DAMP production during viral infection potentiates inflammatory responses through cell-cell communication. Now, we are interested in understanding how DNA DAMPs traffic from infected epithelial cells to patrolling immune cells to activate PRRs, as the molecular and cellular processes governing this intercellular inflammatory circuit are unknown. In combination with CRISPR/Cas9 gene editing and confocal microscopy, we are investigating these questions using a primary human co-culture system that Dr. Barnett developed to study epithelial-immune interactions during SARS-CoV-2 infection.
RNA viruses, such as SARS-CoV-2 and influenza A virus (IAV), infect multiple sites in the body, including the respiratory tract and the gastrointestinal tract. Expression of PRRs varies between organ sites and cell types, meaning that the innate immune response to viral infection may differ based on cell type. Ongoing research to characterize the in vivo effects of PRR activity in murine models of SARS-CoV-2 infection indicate that distinct PRRs contribute to the control of infection at different organ sites. Moving forward, we plan to explore the mechanisms governing activation of these PRRs using primary human organoids and murine models and to expand our investigations to additional sites of infection.
Viral infection fundamentally alters the state of the host cell. This includes not only viral manipulation of host cell protein translation and intracellular trafficking processes but also through alteration of the cellular lipidome and metabolome to support replication. This means that viral DAMPs can take on any of these molecular modalities, and indeed, lipids, nucleic acids, metabolites, and proteins can all activate PRRs. Ongoing work in our lab is characterizing the viral DAMP landscape released from infected primary human airway epithelial cells using unbiased multi-omics and machine learning approaches. Identification of DAMPs associated with different SARS-CoV-2 VOCs will be used to explore the relationship between DAMP release and disease progression.
Viral infection manipulates host cell processes to promote viral replication. This includes innate immunity and cell death, which can have profound effects by either fostering viral replication by enhancing specific cellular activities or restricting viral replication through cytokine production or loss of cell function. Ongoing work in our group indicates that SARS-CoV-2 replication hijacks innate immune pathway function to promote host processes that benefit replication.
Similar to SARS-CoV-2, many respiratory RNA viruses, such as IAV, respiratory syncytial virus (RSV), and other betacoronaviruses, cause disease with varied presentation between infected individuals, including fatal viral pneumonias linked to uncontrolled inflammatory cytokine release. We are interested in understanding if DAMPs also play a critical role in the inflammatory processes mediated by these and other viruses and how these processes differ between viral families and viral disease states.