Data Retrieval

Detailed information for retrieving your data.
Getting Your AGC Data
The University of Michigan Advanced Genomics Core generates and distributes over 800 TB of data a year, and that number is growing. We’ve made a lot of changes in the past few years as we’ve grown from data sets in the KB-range (Sanger) to data in the TB-range (Illumina NovaSeq). Our average data delivery package is now (summer 2026) over 519 GB.
There are two important points for our clients to consider as genomic data grows:
- You are going to get a LOT of data.
- You need a place to store it.
It is your responsibility to get your data and keep it safe. The AGC deletes older data at regular intervals, so you need a plan to receive and store all of your data. To help you, we’ve partnered with U-M’s ITS and ARC, and Michigan Medicine’s HITS to get you the storage and support you need when dealing with many terabytes of data. Check out the section on the Where To Keep Your Data below to get started.
With the sheer volume of data that we generate and move, our old data delivery methods (email, ftp, and external drives) are no longer feasible. We now deliver all data to all our clients using the university-supported Globus file-sharing system.
New Data Distribution Policy
Beginning October 2024, the Advanced Genomics Core (AGC), a unit of the Biomedical Research Core Facilities (BRCF), will only share data with the service request submitter ("Researcher") and the principal investigator for the lab that initiated the service request ("PI").
The AGC policy update follows the Standard Practice Guide policy on Research Data Stewardship (SPG 303.06), which covers the storing and sharing of research data. Get more information on Research Data Stewardship policies that govern the AGC.
How AGC Shares Data
There are two ways that AGC can share data with U-M labs:
- Globus Push: Using Globus File Transfer, the AGC uses a "Globus Push" to a Globus collection that has been previously shared with the AGC. Refer to the “Where to Keep Your Data” webpage on the BRCF website to learn more. This method provides the most flexible, robust, and secure mechanisms for PIs, Researchers, and collaborators. The AGC will continue to announce that the data has been “pushed” to the PI, the Researcher, as well as a list of emails that are included in the original service request submission form.
- Globus Temporary Read Access Link: A "Globus Temporary Read Access Link" can be used by PIs and Researchers. This method sends an email to the PI and Researcher, so they can manually copy the data files to the Globus Collection of their choice over two weeks. It requires action on the part of the Lab, and that’s why the AGC does not recommend this method.
Impact to Labs
Note that this policy update primarily impacts labs using the “Globus Temporary Read Access Link” data delivery method, since the AGC is no longer allowed to grant file access to others, including bioinformatics groups and collaborators.
For labs that need to share files with collaborators, we suggest you set up a Globus collection (the U-M Research Computing Package [UMRCP] is a great place to use as the “backing store” for this collection). You can then direct the AGC to push your data to that collection, and subsequently use Globus to manage sharing data files in that collection with others.
Clients external to U-M should contact the AGC to arrange for data delivery before the completion of their project.
AGC Data Retention Policy
It is your responsibility to keep your data safe, secure, and backed up.
We allow 2 weeks from the time we notify you of its availability in MiCores data to download your data. If the AGC pushed your data to your Globus endpoint, we will keep a copy of your data on our servers for 2 weeks while you ensure it is all there. The best way to ensure you have a complete copy of all of your data is to use the supplied md5 file; see our FAQs below for details on how to use a checksum file.
You must download and keep the entire directory that is shared with you. Although you may not need the raw files for your internal processing, they are commonly required for publishing. Best practice is to keep all of the files you get from the AGC on Data Den, which is why beginning in Fall 2021, the AGC prefers to deliver data directly to your Data Den allocation from the UMRCP. If you have collaborators, bioinformaticians, and others who analyze your data for you, that does not mean that those people will keep a copy of your data forever. You must ensure you have a safe and secure copy of your data. Again, Data Den from UMRCP covers this for you.
After 2 weeks, we archive some of your raw run data, and we keep that archive for 6 months. At your request, we can pull archived data back, although there will be a charge for that service.
We cannot commit to having any of your data after 6 months. Depending on our current rotation policies, we might be able to recover your data after 6 months, but we make no guarantee. If we can recover the raw data, we may need to reprocess it. The minimum charge for this recovery and reprocessing is $241 as of May 2026. If your run is especially complex to recover, or you require additional processing (e.g., cell ranger, space ranger, guppy/dorado, RNA-Seq, etc.), there will be additional charges assessed.
Where to Keep Your Data
External Clients
If you are an external, non-U-M client, we will make your data available for you to download (pull) from our Globus endpoint. This requires you to have a Globus account and a Globus endpoint that can receive the data as you pull it. We will grant read access to our Globus endpoint to the submitter’s email address, as well as the PI’s email address. As stated in our data retention policy above, you will have 2 weeks to pull your data before we delete our copy. If you need more time to get things set up, contact us at [email protected], and we will work something out.
Michigan Medicine "Push to Turbo" Clients
Several of the AGC's clients in Michigan Medicine were already using this “push” data delivery model, but had it set up to go directly to their Turbo allocation. We’ve revised our recommendation and now suggest that all of our clients get their data delivered directly to Data Den. By having the AGC send data directly to Data Den, the lab no longer needs to remember to copy their files to Data Den. And with Data Den allocations at 100 TB (versus 10 TB or less for Turbo), having the AGC push directly to Data Den means it’s less likely that you’ll run out of space. And when you do run out of space, buying additional Data Den storage is 87% cheaper than adding Turbo space.
If your lab is already having AGC push to your Turbo, we recommend following the instructions above to create a new endpoint in your Data Den allocation, and then deleting your Turbo Globus endpoint. (Don’t delete the files; just remove the endpoint from Globus). If you need help, we can help you, and so can the nice folks at ARC.
My Data is on Data Den. Now what?
The good news is that your data is safe. The bad news is that you can’t directly work with files on Data Den, which is, after all, a tape-based long-term archive system. The OK news is that it’s OK – you can’t really do anything with most sequencing files on your PC anyway.
All of our clients will need to do additional processing of the files from the AGC, and all of that additional processing needs to be done on a Linux-based cluster, like the Great Lakes HPC. So, the first step is to copy the files off of the backup tape (Data Den) and put them on high-performance drives (Turbo) for analysis. ARC offers a few options for moving files from Data Den to Turbo, and we prefer using Globus. You’ll need to work with ARC to make sure that your Turbo space is configured for Globus access, and then just use the Globus UI to move the files from your Data Den area to your Turbo area. Then you can fire up Seurat or other analysis packages on the Great Lakes HPC. Teaching you how to move files and run bioinformatic analysis packages is beyond the scope of this web page, but ARC has a set of videos to help you get started.
There are some files that the AGC delivers that are useful on a laptop or workstation. Generally small HTML files and a few other special metadata files. To view those on your laptop, you’ll need to use Globus Connect Personal to download just those files from Data Den to your laptop. We don’t recommend downloading all of the files to your laptop; it will fill your hard drive in a hurry. More info to come in this space – When Data Den updates to Globus V5, the “Download” button may get turned on.
Pulling the Data Yourself with Globus
If your lab wants to pull the data themselves, we can do that. We don’t recommend it because it doesn’t get your data to a safe long-term storage location ASAP. Globus requires some investment of time to use, including creating accounts and ensuring you have an “endpoint” to receive the data files from the AGC’s servers. An “endpoint” is simply the Globus term for “file share”. That is, an “endpoint” is a location to store files that Globus can read and write. You may also know of these as “Network Drives”, “Shared Drives”, or “Mount Points”. We have created a short document to help you get started. Globus’s help section is also a good resource, as is U-Mich’s ARC-TS.
A.) Submitter Pull (this is what external clients need to do)
- This is what you get if you don’t specify a Globus Group or an endpoint that the AGC can write to.
- When your data is ready, we will put your data in our temporary storage area and grant only the submitter and the PI read-only access. You will be notified by a comment in your MiCores submission.
- The PI or submitter must log in to Globus and transfer the data to your Globus endpoint. You have 2 weeks to copy the data off of our endpoint before we remove your data from our file server.
B.) Group Pull (gives your lab direct control over who can download your data)
- Your lab creates a Globus Group with the identities of all Globus users who will be able to copy your data from the AGC’s temporary file server. Important: make sure to allow the group to be viewable for “all Globus users”. This doesn’t make your data public; it just makes the name of your group public.
- When filling out your MiCores submission, enter your group name into the field “Additional Globus ID or Unique Name”
- When your data is ready, we will put your data in our temporary storage area, grant that group read-only access, and notify you via a comment in your MiCores submission.
- Someone from that group must log in to Globus and transfer the data to your Globus endpoint. Your team will have 2 weeks to copy the data off of our endpoint before we remove your data from our file server
Storing Genomic Data on Your PC
We (AGC, BRCF, ARC, ITS, and HITS) do not recommend storing your full genomic datasets on your laptop, pc, lab workstation, or even lab server. Data Den, Turbo, and Locker are the right places for genomic data files at U-M. If you insist on keeping your data elsewhere, you’re on your own.
Pulling Small Files From Data Den to Your PC
You may want to download a few of the smaller run summary files to look at on your PC or laptop. If you want to get those files to your PC, you will need to use Globus Connect Personal, which allows your Windows or Mac computer to create its own Globus endpoint. Globus.org has quite a bit of help available, including a video about using Globus Connect Personal.
Be careful about downloading all of the files for a run, especially .fastq, .bam, and other very large files. The large files aren’t very useful on a PC/laptop since there are no tools available to manipulate them on Windows or Mac OS. Those files belong on an HPC, where they can be properly analyzed and manipulated with bioinformatic toolkits. In the case of data delivered by the AGC, you will only want web (.html) files, loupe browser files (.cloupe), and other sub-gigabyte files. You should select those files individually using the Globus UI to transfer them to your PC. If you blindly download your entire run, you will probably fill your drive with data that you can’t use anyway.
Where to Keep Your Data: U-M Clients
Setting Up Globus
ARC rolled out the UMRCP program for nearly all researchers at Michigan, across all campuses. The U-M Research Computing Package includes free data storage in Turbo, archival storage in Data Den, and Great Lakes HPC cluster usage time. Since this program is free and available to nearly all U-M users of the AGC, we strongly recommend its use, and are building UMRCP into our standard data delivery processes.
Our current best practice is to deliver genomic data directly to our clients’ Data Den allocation. This requires a few one-time configuration steps that your lab must do using Globus after your lab has signed up for the UMRCP. Specifically, you must create an area in your Data Den allocation where the AGC can write your data.
- Log in to Globus.org with full read/write/administrative access to your Data Den allocation.
- Navigate to your Data Den allocation in the Globus File Manager.
- Usually, the collection is UMich ARC Data Den Non-Sensitive Volume Collection, and the path is /school–pi_uniquname.
- For example, /umms-mcgonagall, or /lsa-snape
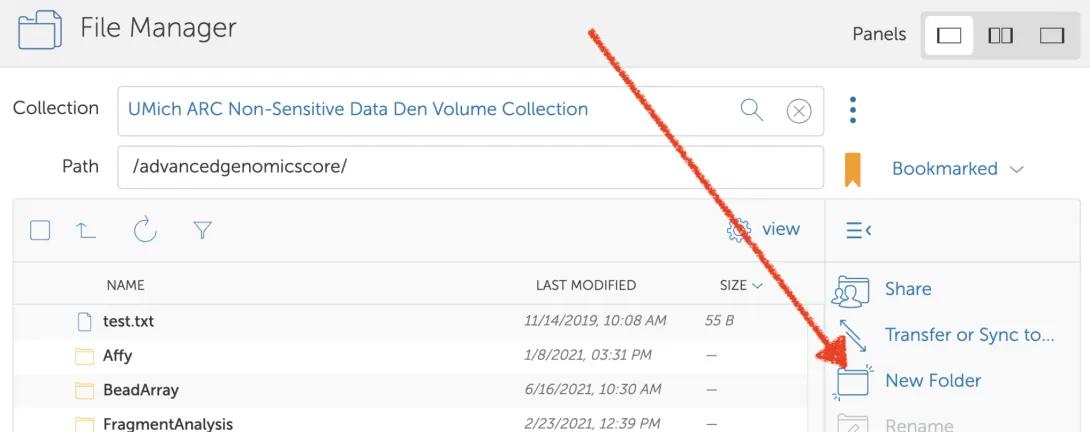
- Create a directory that will receive data from AGC.
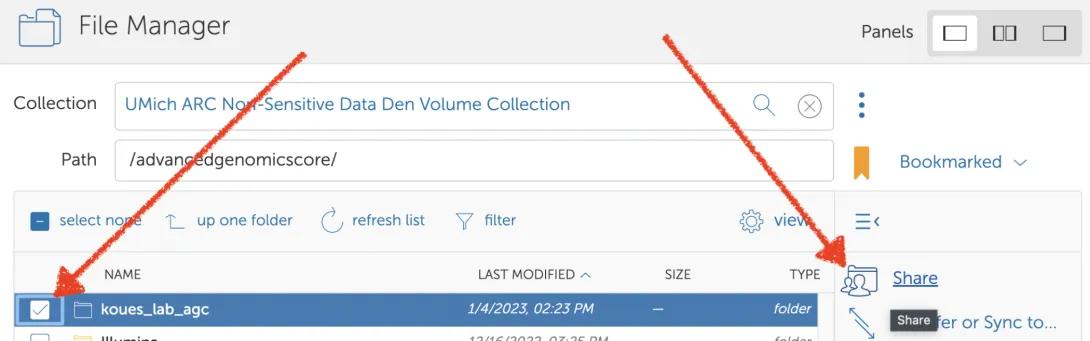
- Select the new folder, and then click “Share”.
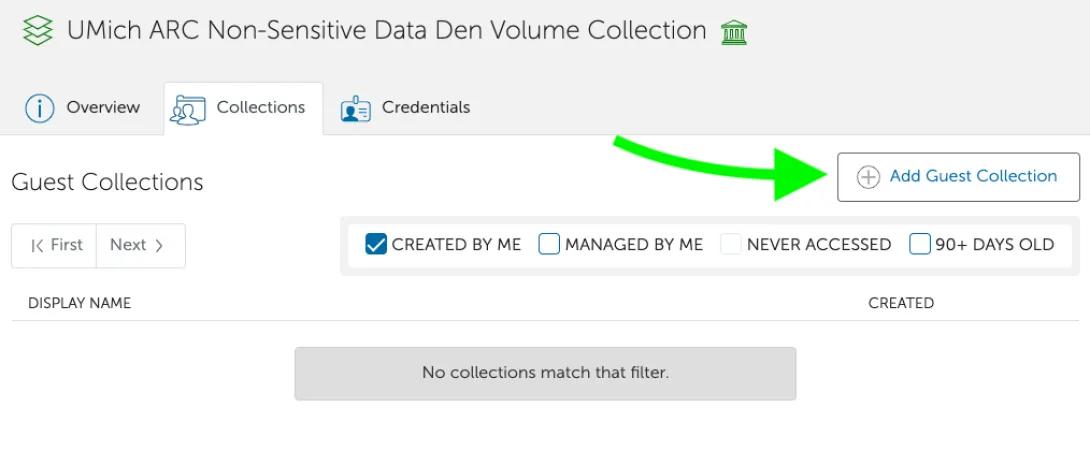
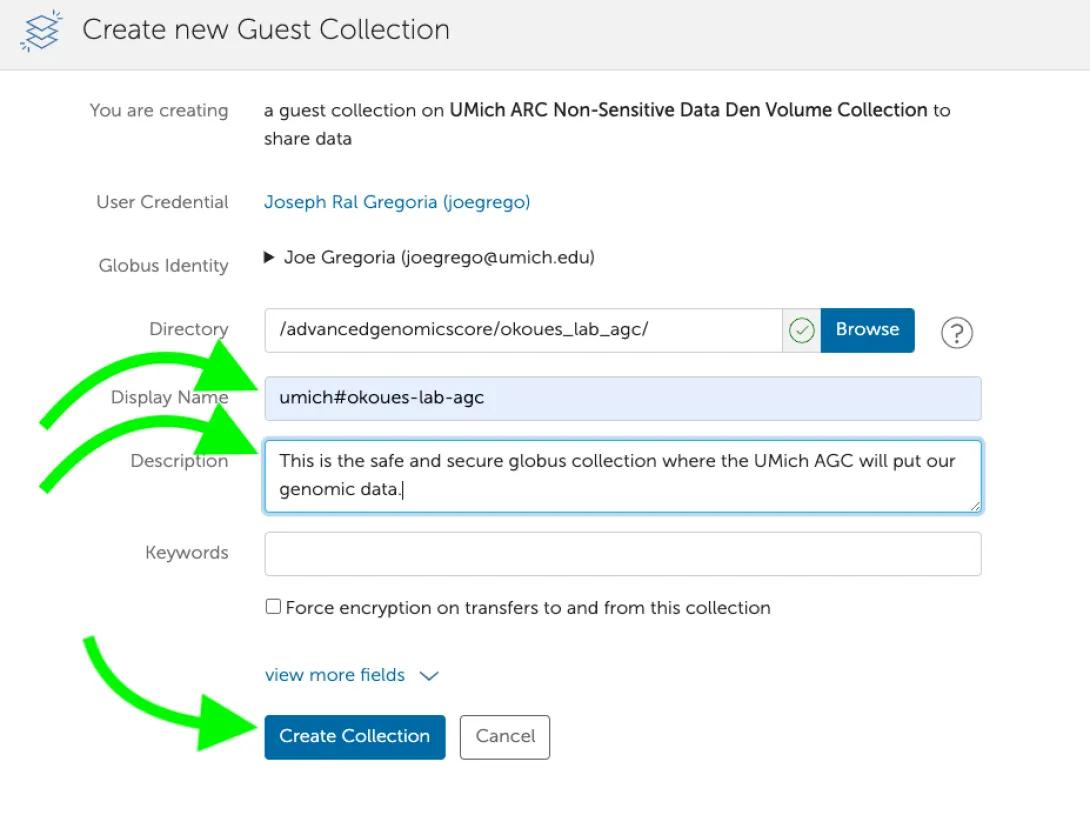
- Click “Add a Guest Collection”
- Fill in Share Display Name (we recommend the form umich#pi_uniqname-Lab-AGC, but that form is not required) and a Description. Click “Create Collection”.
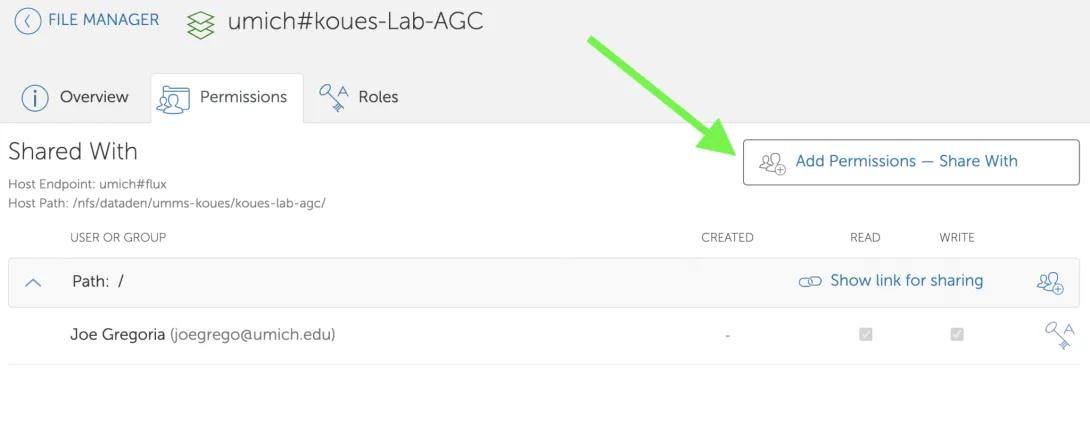
- Click “Add Permissions – Share With”
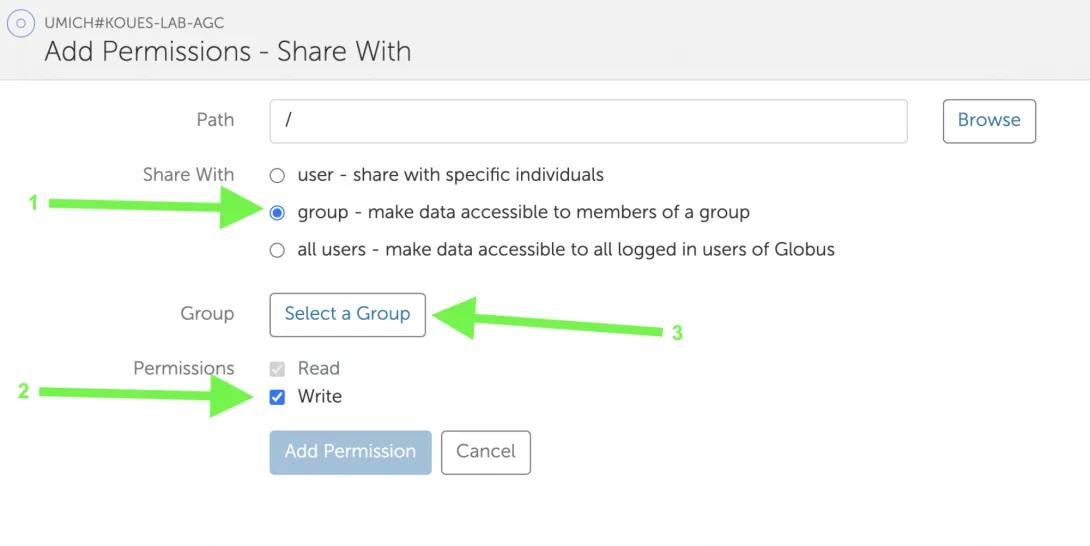
- Select “group” for “Share With” (the default is user, but you need to share with a group instead). Tick “Write” in the “Permissions” section. Then click “Select a Group”.
- In the Select a Group screen, type UMICH-AGC-Data. Click on the drop-down to select it.
New for 2025! The AGC's Globus Group now has an "institution icon" next to it to make it easier to find our official group.

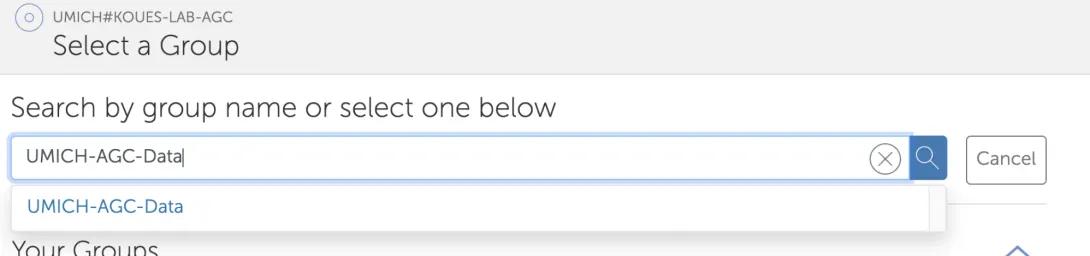
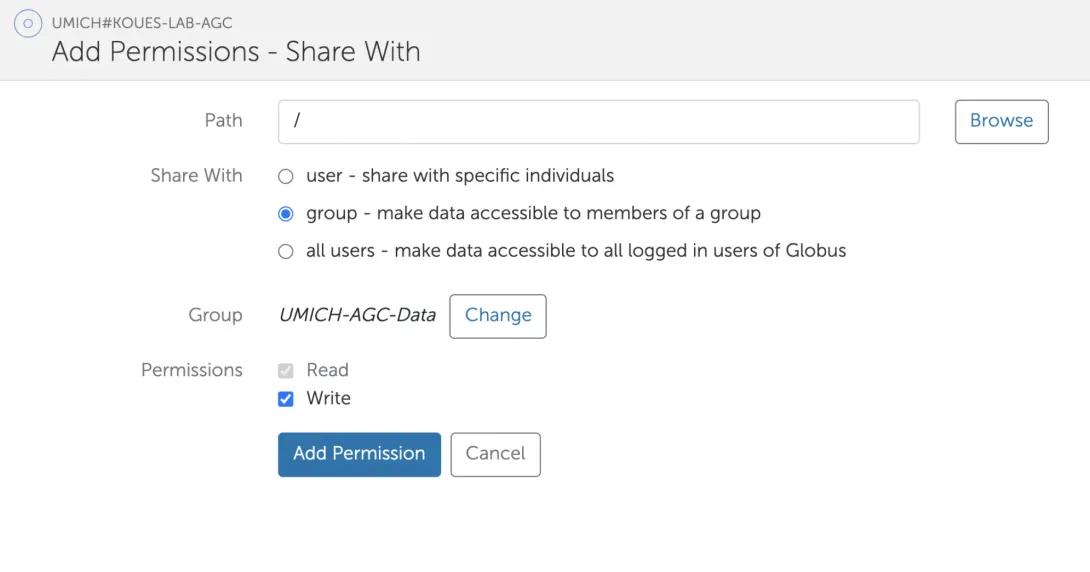
- The “Add Permissions – Share With” screen should look like this. Click “Add Permission” to finish.
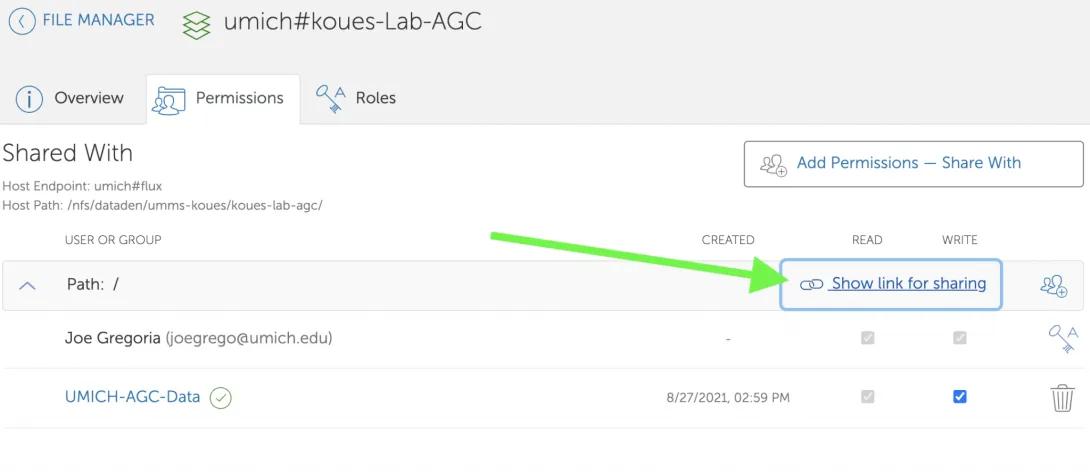
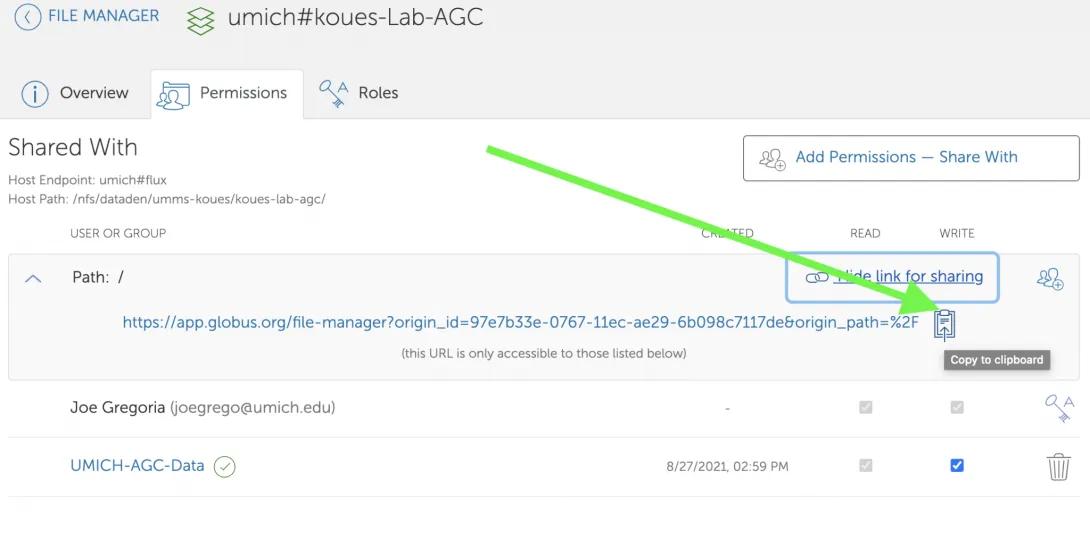
- Click on “Show link for sharing”.
- Click on the “Copy to clipboard” icon to copy your shared endpoint and path.
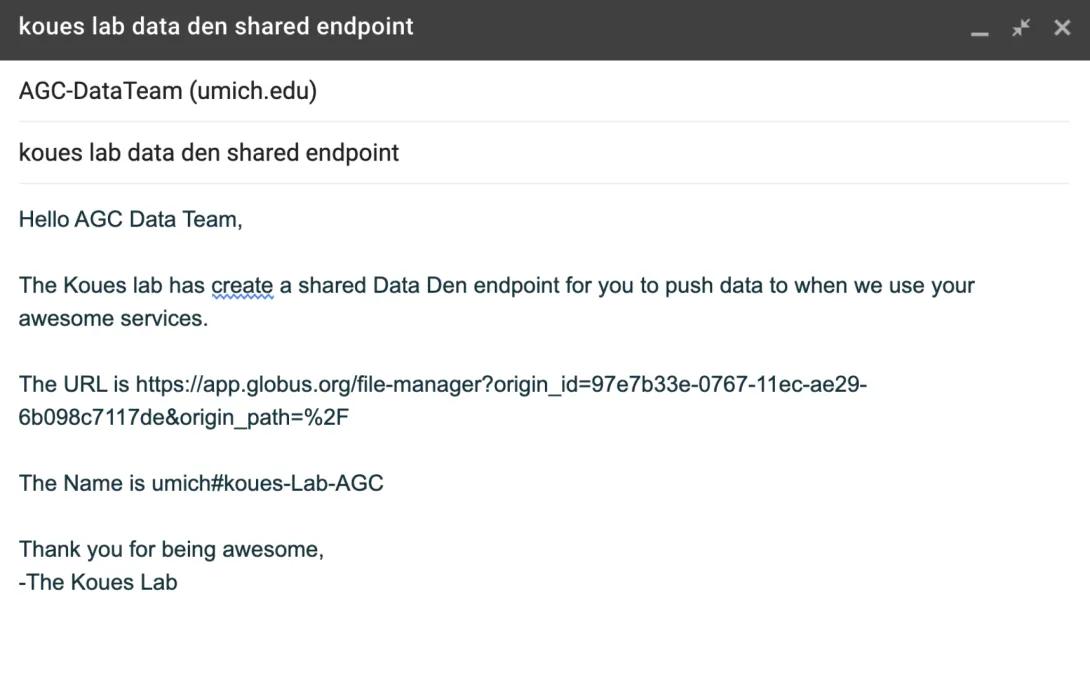
- Create an email to [email protected]. In the subject, tell us that this is your lab’s Data Den shared endpoint. Paste the URL from the screen above. Also let us know the name of this endpoint (in our example above, that would be umich#koues-Lab-AGC). We’ll update our system so all future genomic data pushes from the AGC will go to that directory.
Frequently Asked Questions
What data will I receive from Illumina sequencing?
You will receive demultiplexed fastq files (gzip compressed), containing only the reads from clusters passing the Illumina quality filter. By default, we do not trim the sequencing data. Unless you purchased a full flow cell for yourself, we are not able to deliver fastq files associated with the unknown bin, nor raw bcl files.
How is my data structured?
The following files will be delivered with your data download:
- md5 – Contains checksum information which can be used to confirm the integrity and completeness of the data transfer.
- DemuxStats – demultiplexing metrics and a summary of how your samples performed (this can be opened in Excel). This file is identical to the file we attach to your MiCores submission at data release.
- fastqs – Stored within a directory labeled fastq_[request-id].
- analysis – If additional analysis is performed, a directory labeled with the type of analysis and the request id will be included. This directory will contain, for example, the results of the 10x Genomics cellranger pipeline, RNAseq pipeline, etc., for each sample
- README – Text file containing information about how samples were processed in the lab, information about the sequencing run, and details about the data processing that may be useful for publishing.
Example of the delivered file structure:
123-SR
├── 10x_analysis_123-SR
│ └── Sample_123-SR-1_AAAAAAAA-TTTTTTTT
├── 123-SR.md5
├── DemuxStats_123-SR.csv
├── fastqs_123-SR
│ ├── 123-SR-1_AAAAAAAA-TTTTTTTT_S1_L000_R1_001.fastq.gz
│ └── 123-SR-1_AAAAAAAA-TTTTTTTT_S1_L000_R2_001.fastq.gz
└── README_123-SR.txt
How do I know I have a complete copy of the data?
The first step is to use Globus. A Globus transfer will not succeed unless every file has been successfully moved. Internally at the AGC, we always check the “verify file integrity after transfer” in Globus’s “Transfer & Sync Options” whenever we move files with Globus.
Secondly, data delivered from the AGC includes an “md5” file. This file contains checksums that can be used to confirm that you have a complete copy of every file after the transfer is complete. The checksum file is usually named “<service-request-name>.md5“; an example would be 3092-AG.md5.
Thirdly, if your files are on Data Den, then you will first need to move them to a storage area where you can more easily access the data. We suggest moving the files to Turbo, which is accessible from the Great Lakes HPC cluster. Then, you’ll be able to use a Great Lakes login node to navigate to that Turbo space, for example, cd /nfs/turbo/umms-snape/directory-for-stuff-from-agc/.
Once you have the files in a place where you can access them, you’ll use a command to loop through the contents of your .md5 file and ensure that you have a complete copy of each file. Every operating system has a different command to confirm the checksums. On most Linux-based systems, like the Great Lakes HPC, you would use a process like this:
- cd <service-request-name>
- md5sum --quiet --check <service-request-name>.md5
- Pay attention to any output; the --quiet flag means that the md5sum program will only generate output if there is a problem.
Remember that the AGC only makes your data available for 2 weeks, and that after 6 months our copy of your data is destroyed. We strongly encourage all clients to download their data as soon as possible, and use the provided md5 file to verify each download as soon as possible. If there are issues, a second Globus transfer of the entire directory usually clears up the issues. If you still see checksum issues after re-trying the transfer, contact the AGC.
What data will I receive for Single Cell projects?
- Fastq’s – We use bclconvert to demultiplex all sequencing data. You will notice that each sample will have 4 fastq directories associated with it (one for each of the 4 barcodes in the 10x barcode set for each sample), named Sample_[number]_[letter]. You will find the normal output (demultiplexed fastqs) in the following path: [run_id]/[service_request_id]/Sample_[number]_[letter]
- Cellranger count output – We run CellRanger Count on all single cell gene expression samples. Inside the top directory of your download is a directory for each sample by name that contains the results from the count step of the cellranger pipeline. The web_summary.html is likely what you want to look at first. The cloupe.cloupe file can be opened in the Loupe Browser supplied by 10x. You will be able to find this file in the following path: [run_id]/Sample_[name]/outs
- If your samples are not standard single cell RNAseq, we will run the appropriate supported 10x software for your project type
- We do not support processing of TotalSeq-A projects. You will only receive demultiplexed fastqs.
Please contact us if you would like assistance interpreting the results produced by cellranger. We will do our best to answer any questions, or we can guide you towards assistance and resources provided by 10x.
What data will I receive from an Oxford Nanopore project?
Your data package for an ONT run will include:
- an MD5 file to validate your download
- the ION instrument run output (fast5/pod5 files, etc.) for each run
- Guppy basecaller outputs (fastq.gz files, sequencing_summary.txt, sequencing_telemetry.js)
- pycoQC* basecalling metrics and summary report(s)
Check out the guide to reading the pycoQC report.
What part of my data should I download?
You need all of the files. You also paid for them all. And it will cost you hundreds to thousands of dollars to recover them, if they are even recoverable, when you actually need them to publish.
(FWIW, about 4 times a year, the AGC tells someone who can't find their files that their FASTQ files are gone really gone for good. We really, really don't keep backups for very long. Without their fastq files, the researcher can't publish their findings. It is very sad. Don't be that researcher.)
What if I need additional sequencing for my samples?
What if I need Bioinformatics Help?
An initial NGS project consultation with the U-M BRCF Bioinformatics Core is free.
I set up an account for data-den, but I am not quite sure how to transfer the data to my account and share it with my collaborators?
We have a lot of information on our website, including links to both Globus and ARC how-tos and videos. michmed.org/agc-data
In a nutshell:
- Set your lab up so that AGC can push data to your Data Den location. michmed.org/agc-data-den-setup
- If you need to move files yourself to Data Den, refer to https://www.globus.org/data-transfer
- Once the files are there, you can use the same process that you used to grant AGC write access to grant "read only" access to either your entire data den repository, or to just specific folders/projects. For another view on that process, see https://www.globus.org/data-sharing
ARC provides lots of information, videos, and even one-on-one coaching to labs who are using Globus. Reach out to them to get more personalized help. https://its.umich.edu/advanced-research-computing
What will I get from an RNA-Seq project?
Introduction
For certain RNA-Seq projects, in addition to FASTQ files, the AGC can provide (at no additional cost):
- aligned reads
- preliminary QC results
- a table of gene read counts (suitable for use in differential expression analysis, although the AGC does not provide differential expression analysis)
These additional data files will only be generated for bulk RNA-Seq experiments using poly-A or total RNA library preps, and only for specific organisms. We generally support eukaryotic vertebrates and most other model organisms. Usually we can accommodate species listed in this Ensemble page, if you communicate with us before your project has begun: https://useast.ensembl.org/info/website/archives/assembly.html.
We use the nfcore/rna-seq pipeline to do this analysis, and so are additionally limited by the capabilities of that pipeline.
If you need additional analysis or support, the Bioinformatics Core ([email protected]) can provide the additional downstream analysis (e.g., differential expression modeling, pathway/gene enrichment analysis) on these or other RNA-Seq experiments.
Pipeline Details
Starting in July 2025, the AGC uses the nf-core/rnaseq pipeline for our RNA-Seq projects to perform common tasks such as QC, trimming, alignment, and quantification.
You can find more details about the NF-Core pipeline at the following link: https://nf-co.re/rnaseq. There you will find details on the outputs, the process, and many additional details about this pipeline developed by the nf-core community.
We have added a handful of files to these outputs to provide additional information to our clients:
- star_rsem/rsem.merged.gene_counts.annot.tsv
- An annotated version of the gene expression count matrix provided by rsem, where annotation information has been obtained from BiomaRt.
- pipeline_info/ref_source_info.yaml
- Useful information about the reference files that were used and where they can be downloaded
- pipeline_info/params_{project}.yaml
- A parameters file that mirrors what was used for this analysis
- The parameters file has some sections where the text "/path/to/" is a placeholder for the actual path to the resource
- pipeline_info/samplesheet_{project}.csv
- The samplesheet that mirrors what was used for this analysis
- The samplesheet has some sections where the text "/path/to/" is a placeholder for the actual path to the resource
History
Between 2020 and 2025, the AGC used an in-house developed pipeline for RNA-Seq; we now use the open source "nf-core/rnaseq" pipeline (https://nf-co.re/rnaseq). The nf-core/rnaseq pipeline uses the same component tools as our old pipeline and produces equivalent results. The new pipeline has been broadly adopted in bioinformatics; it is extremely well supported in the open source community, and it executes more efficiently in the U-M computing environment.
If you used us before for RNA-Seq, the outputs that you see now will be somewhat different than what you received in the past. Basically, all results from the NF-Core pipeline are included, and some additional files are added to provide helpful information or include artifacts important for reproducibility.
Our most common questions regarding this change have to do with TPM and FPKM count files.
The unannotated raw count matrix and TPM normalized count matrix are available in the standard nf-core pipeline results, under the "star_rsem" directory; see https://nf-co.re/rnaseq/output#star-via-rsem for more info on outputs from nf-core/rnaseq.
The FPKM normalized count matrix is not included in an aggregated form, but all of the information is available in the individual {sample}.genes.results files if you want to aggregate them yourself. We provide an annotated version of the most useful count matrix - the gene-level non-normalized expression count matrix - which would correspond to our old pipeline's "gene_expected_count.annot.txt" file. This can be found at "star_rsem/rsem.merged.gene_counts.annot.tsv". This is one file that we manually generate in addition to the standard nf-core results, because the annotated version of this file is most useful for checking out results or performing downstream steps. It has annotations added from BiomaRt - these annotations are often very helpful to researchers when scanning through the results, since gene symbols & descriptions are recognizable compared to their unique ENSEMBL IDs.
Note: If your analysis requires running the legacy pipeline on your data, you can contact the Bioinformatics Core to set up a project consultation.
What files will I receive with a Load & Go run?
- No Demultiplexing or Data-level QC: For "Load & Go" projects, we deliver files exactly as they are generated by the sequencers. Demultiplexing and data-level quality control will no longer be performed for Load & Go projects. Your lab may need to process the sequencer output using software such as Illumina’s bcl-convert.
- Globus Push Required: To keep turn-around time as fast as possible for all users of the Advanced Genomics Core’s (AGC) “Load & Go” service, we require that labs using this service set up a Globus Collection with the AGC. For detailed instructions on setting up a Globus Collection, see michmed.org/agc-data-den-setup
- Alternative Options: If you require demultiplexed files, or if you do not wish to set up and maintain a Globus Collection, you can opt to submit sequencing-ready libraries instead and utilize our sequencing-ready library rates.
Can you share my data with...?
- For University of Michigan clients, the answer is "no, we can't". Both U-M and AGC policies prohibit us from sharing your data with someone else. It is your data; storing and sharing your data is your responsibility. There are options for you to download the data and share it, detailed at the top of this page (michmed.org/agc-data), including the Data Distribution Policy, Pulling the Data Yourself with Globus, and Where to Keep Your Data. For additional help, we suggest you contact HITS, ITS, or ARC to set up your lab's storage solution at U-M.
- For non-University of Michigan clients (that is, you and your PI don't have an @umich.edu email address and you paid external rates for AGC services), we can work with you to get your data; contact us at [email protected].
We Cannot Share Your Data
- For UMich clients, the answer is "no, we can't". Both U-M and AGC policies prohibit us from sharing your data with someone else. It is your data; storing and sharing your data is your responsibility.
- For non-UMich clients (that is, you and your PI don't have an @umich.edu email address and you paid external rates for AGC services), we can work with you to get your data; contact us at [email protected].
Questions?
Contact Us
University of Michigan
2800 Plymouth Rd.
Ann Arbor, MI 48109-2800