Transgenic Core Laboratory Protocols
See specific protocols below.
Quick Links
Laboratory Protocols
We have found the NucleoSpin® Extract II Kit (available from Macherey-Nagel, Catalog number 740609.50) is a simple and fast way to obtain microinjection quality DNA. This is not to exclude other methods of DNA purification but to say that we find this is a convenient method that is less time consuming than CsCl gradients.
- Perform restriction digest to liberate transgene from plasmid vector sequences. Final yield should be 10 – 20 micrograms of transgene insert.
- Separate restriction digest products on agarose gel using either TBE or TAE. Use either low- or standard- melting temperature agarose.
- Place gel on transilluminator. Cut out band(s) of interest. Use a clean razor blade or scalpel. Remove as much excess agarose as possible. Minimize DNA exposure to UV light to prevent photochemical damage (less than 1 minute).
- Transfer agarose slice(s) to a preweighed tube. Reweigh tube to determine weight of agarose in tube.
- For each 100 mg of gel, add 200 microliters buffer NTI. If agarose concentration is greater than 1%, add proportionately more buffer. For example, if a 2% agarose gel is used, add 400 microliters buffer NTI for each 100 mg of gel.
- Dissolve at 50 degrees Centigrade for 10 minutes, vigorously vortexing every 2 to 3 minutes until the agarose is completely dissolved.
- Place a NucleoSpin cartridge in a 2 ml micro tube and load 750 microliters dissolved gel slice onto the cartridge. Spin at 11,000xg for 60 seconds in a microcentrifuge. Discard the flowthrough. The cartridge has a capacity of 15 micrograms DNA, so you can run several 750 microliters loads of dissolved gel slice through a single cartridge.
- Filter 750 microliters of buffer NT3 through Anotop 10 0.02 micron filters (GE Healthcare Life Sciences, Catalog number 6809-1002). Add buffer to the cartridge and spin at 11,000xg for 60 seconds in a microcentrifuge. Discard the flow-through.
- Replace tube with a fresh micro-tube. Spin the empty cartridge at 11,000xg for 60 seconds in a microcentrifuge to completely remove buffer NT3.
- Elute the DNA from the cartridge: Replace tube with a fresh micro-tube. Add 50 microliters of preheated (60 degrees Centigrade) elution buffer to cartridge and incubate one minute. Spin at 11,000xg for 60 seconds in a microcentrifuge.
- Elution buffer: 10 mM Tris-HCl, pH 8.5, 0.02 micron filtered. Check the pH of the elution before just before you use it, best yields are obtained at a pH of 8.5 or greater. The 0.02 micron Anotop syringe filters are available from Whatman (GE Healthcare Life Sciences).
- If you have problems with particulates plugging the micoinjection needles, pre-filter the wash and elution buffers with the 0.02 uM filters. Do not filter the DNA through the filters, the small pore size will trap DNA molecules.
- If desired, repeat step 10 to increase yield. We obtain 90% of the DNA in the first elution.
- Quantitate DNA solution.
- Verify size and intact condition of DNA on minigel.
- Store eluted DNA at -20 degrees Centigrade.
BAC DNA has been purified successfully for microinjection by three principle methods: Anion Exchange Chromatography (Nucleobond Columns), CsCl Gradient, or Sepharose 4B-CL Chromatography. Any of these methods can be used successfully to purify BAC DNA for pronuclear microinjection when the protocols are followed carefully.
Nucleobond Column Method
This method was established in the University of Michigan Transgenic Core for our users. The method is used to purify BAC DNA for microinjection into fertilized mouse eggs and BAC transgenic mouse production.. The following method yields clean, non-sheared DNA. Some of information below was taken from The Nest Group web site (The Nest Group, Inc. TEL 800-347-6378, FAX 508-485-5736).
Materials
The kits are no longer available from The Nest Group, but can be purchased from Clontech (Takara Bio).
NucleoBond BAC 100 Kit, Clontech Cat. #740579 This gives the best compromise of volume management and size (capacity). This kit contains 10 AX-500 cartridges.
NucleoBond Buffer Set I, Clontech Cat. #740601. Without the Buffer Set I you will have to make up more of the S1, S2, S3 buffers since there is not enough in the PC kit to double the volumes of extractants of a normal plasmid, which is necessary to do BACs.
NucleoBond Folded Filters are included in the BAC 100 kit. The filter papers will eliminate the centrifugation step after precipitation of the cellular debris and will clarify the extraction in about 10 minutes instead of a 45 minute spin. This will reduce the exposure time to nucleases as well as reduce the shearing potential of a centrifugation. This is highly recommended.
Special Conditions for Purifying BACs
When isolating BACs with the Nucleobond AX alkaline lysis-based protocol, the potential always exists for lower than expected yields to be obtained. While, in general, there are a number of factors that could adversely affect plasmid yield, in the case of low copy number plasmids, incomplete bacterial lysis is the number one culprit. The primary reason for this is that in order to get the maximum yield for a particular cartridge size, many researchers grow cultures of these plasmids that are larger than can be efficiently handled by the prescribed volumes of buffers for the cartridge. The first and most obvious solution is to increase the amounts of the buffers to be used in these cases to reduce viscosity and to promote diffusion.
- (A) Some Golden Rules for Lysis Buffer Volumes
Use the following minimum volumes of the cell-suspension/RNAse buffer S1, lysis buffer S2, and high salt co-precipitation buffer S3: Use a minimum of 4.0 ml of each buffer per 100 ml of culture regardless of which cartridge size is being used. However, if the particular cartridge requires more than this, then use the prescribed amount for the cartridge. For example, if a 100 ml BAC culture is to be processed on an AX-500 cartridge then use the prescribed 12 ml of S1, S2, and S3 for the cartridge. On the other hand, if a 500 ml BAC culture is to be processed on an AX-500 cartridge then at least 20 ml ({500 ml / 100 ml} * 4) of each of the three buffers should be used according to this rule to insure proper cell lysis.
- (B) Use of an additional volume of N5 Elution Buffer (see below)
If the elution step is repeated one additional time (see Step 8, in A Modified Alkaline Lysis Procedure for the Purification of Plasmids and Cosmids), up to 30% more DNA can be isolated. This is especially true for the AX-100 and AX-500 cartridges. Note the flow rates of Nucleobond AX cartridges are up to two times faster than for Qiagen Tip cartridges which means that clogging from higher viscosity solutions is less likely (less sensitive to cell density problems), and the amount of losses from endonucleases will be lower due to shorter contact times.
- (C) Use of a Denaturing Elution Buffer
It has been reported in some cases that the use of a high GC content type of elution buffer helps to increase the yields of very large plasmids such as BACs regardless of their GC content. The formula for this elution buffer is as follows: 50% formamide, 1.0 M KCl, 15% EtOH & 0.1 Tris-phosphate, pH 8.5. This buffer should be heated to 60C before loading. For the recommended procedure for preparing this buffer refer to N5 Elution Buffer Preparation (see below). However, use of this formamide buffer can lead to complications in the precipitation step unless care is taken to prevent salt precipitation (room temperature propanol precipitation and centrifugation is required), formamide removal (a second propanol precipitation is required) and propanol removal (an additional ethanol precipitation is required).
- (D) Notes Contributed by Chris Russell, RESEARCH GENETICS, INC: [email protected])
– Some BACs will yield better and more DNA when grown at room temp to 30 C rather than at 37 C. This is not generally true, but some BACs are unable to grow vigorously at 37 C.
– Many BACs will yield better in LB broth than in Terrific broth or other rich broths. It may be a growth problem or a technical problem due the presence of a higher concentration of bacterial cells.
– Concentration measurements by UV spectrophotometry are not very reliable and may not be meaningful, since you can have absorbing contaminants as well as significant amounts of E. coli DNA. Same is true for flourometry due to the presence of E. coli DNA. Same goes for checking the BAC on a regular agarose gel. By pulsed-field gel electrophoresis of Not I digested BAC DNA you can evaluate the integrity, quality, and quantity of BAC DNA.
Modified N5 Elution Buffer Preparation
This modified buffer is designed increase yields of GC rich DNA or other “sticky” DNA from the Nucleobond column. The buffer contains 50% formamide, 1.0 M KCl, 15% EtOH, 100 mM Tris-phosphate, pH 8.5.
- Add 12.11 Tris base to 250 ml distilled water and dissolve
- Add 74.55 g KCl to Tris solution
- Autoclave this solution
- Add 150 of absolute ethanol to cooled solution
- Add 500 ml of formamide to solution
- Adjust pH to 8.5 with phosphoric acid – do not overshoot pH
- Adjust final volume with sterile distilled water to 1000 ml
- Store at room temperature
Method
Using this modified method of Birnboim and Doly, bacterial cells are lysed by a NaOH/SDS solution. Chromosomal and plasmid DNA are partially denatured under these alkaline conditions. It is important to control the duration of this denaturing step thoroughly. The following addition of potassium acetate is a crucial step as well. It results in a precipitate containing the chromosomal DNA and other cellular compounds. Plasmid DNA stays in solution and is purified to homogeneity on the corresponding NUCLEOBOND AX cartridge. For buffer volumes and operation conditions see the below. Please read the remarks before you start your preparation. Use 100-500ml of bacterial culture (A600 approximately 1 O.D.) in conjunction with one AX-500 cartridge.
Cell disruption:
- Carefully resuspend the bacterial cell pellet in buffer S1: 12 ml (see Note A above)
- Add the appropriate volume of buffer S2: 12 ml (see Note A above) Mix gently by inverting the tube, and incubate at room temperature or 5 min. Do not vortex to prevent the release of chromosomal DNA from the cell debris.
- Add buffer S3: 12 ml (see Note A above), Mix gently by inverting 6-8 times until a homogeneous suspension is formed. Incubate the mixture on ice for 10 minutes to begin the precipitation of SDS and cellular debris. Put the folded filter onto a 50 ml tube or equilibrated Nucleobond AX cartridge, wet it with 0.5 – 1.0 ml of water and fill it with the cooled lysate. Collect the cleared flow-though. These filters are suitable for plasmid isolation with Nucleobond AX 100 and larger cartridges, where the volume of the lysate is sufficient to effect a complete hydration and wash of the filter paper. To get the maximal recovery of DNA, rinse the filter paper with an additional 1.5 ml of water.
Equilibration
Equilibrate an AX-500 cartridge with 5 ml buffer N2
Adsorption
Load the collect the cleared flow-though from the filter paper onto an equilibrated an AX-500 cartridge.
Wash
Wash cartridge with 2×12 ml buffer N3.
Elution
Elute DNA with 6 ml buffer N5 (see notes B, C, and Modified N5 Elution Buffer Preparation above). If this elution step is repeated one additional time, up to 30% more DNA can be isolated. This is especially true for the AX-100 and AX-500 cartridges.
Precipitation
Precipitate DNA by adding 0.7 volumes of room temperature iso-propanol. Centrifuge at >12000 x g at 4C for 10-20 minutes. Wash the pellet with 70% ethanol, air dry briefly (about 5 minutes), and dissolve in microinjection buffer (see below).
Resuspend DNA
Resuspend DNA in 100 ul microinjection buffer (see below) available from the Transgenic Core.
Quantitation
Check concentration by 0D 260/280. We and others have observed that there is no difference in efficiency of producing transgenic mice with circular BAC DNA or linearized BAC DNA. You may submit either form of DNA for microinjection. When you do so, also submit 10 microliters of Not I digested BAC DNA for pulsed field analysis. This will allow us to assess the purity of your DNA preparation. We will verify the concentration of your concentrated DNA sample and adjust it for microinjection to 0.5 to 1.0 ng per ul.
DNA Storage
Store resuspended DNA at 4C.
Buffer Storage
- S1 – 50 mM Tris-HCl, 10 mM EDTA, 100 µg RNAse A / ml, pH 8.0, 4C
- S2 – 200 mM NaOH, 1% SDS, RT
- S3 – 2.8 M K-Acetate, pH 5.2, 4C
- N2 – 100 mM Tris, 15% ethanol &900 mM KCl adjusted with phosphoric acid to pH 6.3, RT
- N3 – 100 mM Tris, 15% ethanol &1150 mM KCl adjusted with phosphoric acid to pH 6.3, RT
- N5 – 100 mM Tris, 15% ethanol &1000 mM KCl adjusted with phosphoric acid to pH 8.5, RT
*This salt concentration is sufficient for the elution step, because of the increased pH value of this buffer.
The RNAse (already heat treated and DNAse-free), must be added to buffer S1 before use. S1 should be stored then at 4C. The SDS in buffer S2 will precipitate at temperatures below 20C. If this is the case, store the bottle for a few minutes at about 30C to 40C and mix well and equilibrate to room temperature before use. The SDS is removed by buffer S3 (white precipitate) and will not be loaded on the cartridge. This step is very important! Take care that the supernatant of step 5 is clear! SDS will clog the cartridge and prevent the adsorption of nucleic acids.
Preparation of Microinjection Buffer
Very Important -> Microinjection Buffer In our hands, successful BAC preparations require the polyamine microinjection buffer described below. We have stored intact BAC DNA at 4C for a year in polyamine buffer. |
Buffer composition is 10 mM Tris-HCl, pH 7.5, 0.1 mM EDTA, 30 microM spermine, 70 microM spermidine, 100 mM NaCl. This buffer is more likely to produce transgenic mice with intact, unfragmented DNA molecules. See the following references: Schedl A, Larin Z, Montoliu L, Thies E, Kelsey G, Lehrach H, Schutz G. 1993. A method for the generation of YAC transgenic mice by pronuclear microinjection. Nucleic Acids Res 21:4783-4787 and Montoliu L, Bock CT, Schutz G, Zentgraf H. 1995. Visualization of large DNA molecules by electron microscopy with polyamines: application to the analysis of yeast endogenous and artificial chromosomes.. J Mol Biol 246(4):486-492. See also Lluis Montoliu’s website.
1000x Polyamine Stock
30 mM Spermine (Sigma, tetrahydrochloride, #S-1141)
70 mM Spermidine (Sigma, trihydrocholoride, #S-2501)
Dissolve the spermine and spermidine together in autoclaved distilled water, filter sterilize (0.2 micron filters), and store at -20 C. Since the polyamines are very hygroscopic, it is suggested that small quantities (1 gram) should be ordered and then all of it should be prepared at once.
Microinjection Buffer
For 50 ml:
10 mM Tris-HCl, pH 7.5 0.5 ml of 1 M Tris-HCl, pH 7.5 (autoclaved)
0.1 mM EDTA, pH 8.0 10 microliters of 0.5 M EDTA, pH 8.0 (autoclaved)
100 mM NaCl 1 ml of 5 M NaCl (autoclaved)
1x Polyamines 50 microliters 1000x Polyamines mix
Autoclaved H2O up to 50 ml
NOTE: Prepare fresh and discard unused microinjection buffer.
- Purify Plasmide from Bacteria
- We recommend the Qiagen EndoFree Plasmid Maxi kit for the purification of the targeting vector plasmid from bacteria. Please follow the directions in the kit. Electroporation of Qiagen purified DNA has been used successfully by a number of labs. Alternatively, plasmid DNA can be purified by CsCl banding.
- Linearize 200 micrograms of plasmid DNA with the appropriate restriction enzyme digest
- Run a DNA on a minigel to verify that digestion is complete. Extract the DNA with phenol-chloroform, then with chloroform and precipitate by adding NaCl and ethanol. Make sure you use fresh phenol with neutral pH for maximum DNA recovery and highest cell viability in electroporation. Wash the DNA pellet in 70% ethanol and allow to it air dry. Resuspend the DNA in sterile TE (10 mM Tris-HCl, pH 8.0, 1.0 mM EDTA) at 2 mg/ml and deliver it to the Transgenic Core for electroporation. Prior to electroporation, we will verify the concentration and run it on a minigel to check the size and look for degradation.
Materials
- Blastocysts placed in 10 ul of water in PCR tubes and frozen at -20C or -80C
- 2X Blastocyst Lysis Buffer
- Pipetman
- RNAse Free Pipet Tips
- PCR tubes
- PCR primers for the gene of interest
- Gene specific PCR primers and Taq polymerase of choice
- Thermal Cycler for pCR
- 1 M KCl (Sigma Cat. no. 60142)
- Tween 20 (Sigma Cat. no. P9416)
- 2% Gelatin (Sigma Cat. no. G1393)
- Nuclease Free Water (Sigma Cat. no. W4502)
- 10 mg/ul yeast tRNA (Sigma Cat. no. R5636) store at -20C
- 20 mg/ml Proteinase K (New England BioLabs Cat. no. P8107S) store at -20C
Procedures
Blastocyst Lysis Buffer Base (100 mM Tris-HCl, pH 8.3 100 mM KCl, 0.02% gelatin, 0.45% Tween 20)
For 10 ml, mix together:
- 2.0 ml 1 M Tris-HCl (pH 8.3)
- 2.0 ml 1 M KCl
- 40 ul 2% Gelatin
- 90 ul Tween 20
- 5.87 ml Water
Store at room temperature.
2X Blastocyst Lysis Buffer (add 60ug/ml yeast tRNA, 125 ug/ml Proteinase K)
For 1 ml, mix together:
- 1 ml Blastocyst Lysis Buffer Base
- 16 ul 10 mg/ul Yeast tRNA
- 62. ul 20 mg/ml Proeinase K
Blastocyst Lysis to produce crude DNA solution
Add 10 ul of 2X Blastocyst Lysis Buffer to each blastocyst frozen in a PCR tube
Place PCR tubes in the thermal cycler
Run the following program to prepare the crude DNA solution:
56C for 10 min
95C for 10 min
4C indefinitely
Store crude lysate at -20C
Blastocyst PCR
Use 4 ul of crude blastocyst DNA in a 20 ul PCR reaction to detect the gene of interest.
nota bene; We have had good results with KOD polymerase when other Taq polymerases didn’t amplify.
Protocol adapted from Sakurai T, Watanabe S, Kamiyoshi A, Sato M, Shindo T. 2014. A single blastocyst assay optimized for detecting CRISPR/Cas9 system-induced indel mutations in mice. BMC Biotechnol. 14:69. PMID: 25042988
Results
Highly specific and sensitive assays should be used because most PCR reactions are based on the use of 1 ng DNA per PCR reaction. One blastocyst is about 60 cells; a single cell contains about 6 pg of DNA; thus the starting material for a blastocyst PCR is 4 ul crude lysate X (60 cells X 6 pg DNA/cell)/20 ul crude lysate) or 72 pg DNA per PCR reaction.
Primer Design Suggestions for specific and sensitive PCR assays
Use Primer-Blast to pick primers: http://www.ncbi.nlm.nih.gov/tools/primer-blast
Adjust Primer Parameter default settings
- Minimum primer melting temperature: change to 60°C
- Optimal primer melting temperature: change to 63°C
- Maximum primer melting temperature: change to 66°C Minimum primer melting temperature difference: change to 1°C
Adjust Specificity Checking Parameters
- Click box to turn on “Enable search for primer pairs specific to the intended PCR template” Set Search Mode to “Automatic”
- Set Database to “Genome (reference assembly from selected organisms)”
- Set Organism to “Mus musculus (taxid: 10090)”
Click on “Advanced Parameters”
- Set Primer Size Min to 27
- Set Primer Size Opt to 29
- Set Primer Size Max to 31
Stratman et al. reported primers of 27-30 nucleotides made up of 50-60% GC content and that will produce a 100-500 bp PCR product uniformly detect genomic templates with single copy sensitivity.
Stratman JL, Barnes WM, Simon TC. 2003. Universal PCR genotyping assay that achieves single copy sensitivity with any primer pair. Transgenic Res. 12:521-522. PMID: 12885173
PCR Enhancer Mix
The 5X CES PCR enhancer mix described by Ralser et al. can improve the results obtained from difficult PCR reactions. The authors further recommend that the PCR reaction buffer contain the following final concentrations of components: 65 mM Tris–HCl, 16.6 mM (NH4)2SO4, 3.1 mM MgCl2, and 0.01% (v/v) Tween 20 at a pH of 8.8.
5X CES
2.7 M betaine 6.7 mM DTT 6.7% DMSO 55 ug/ml BSA
Ralser M, Querfurth R, Warnatz HJ, Lehrach H, Yaspo ML, Krobitsch S. 2006. An efficient and economic enhancer mix for PCR. Biochem Biophys Res Commun. 347:747-751. PMID: 16842759
Materials
- in vitro transcribed mRNA or RNA
- Oligonucleotide resuspended in RNAse free water
- DNA donor plasmid prepared with an endotoxin free plasmid kit
- 0.02 µm Anotop 10 Syringe Filters (Whatman Cat. no. 6809-1002) — Do NOT filter nucleic acid solutions through the Anotop filters, the 0.02 µm pores will trap nucleic acids and remove them from solution
- Millipore dialysis filter (Millipore #VMWP02500, pore size 0.05 µm)
- Sterile 10 ml syringe
- Sterile 10cm Petri dish
- Pipetman
- RNAse Free Pipet Tips
- Sterile 1.5 ml Microtubes
- Sterile 15 ml tube
- Sterile 50 ml tube
- Sterile 10 ml pipets
- 1 M Tris-HCL, pH 7.4 (Sigma Cat. no. T263)
- 0.5 M EDTA (Sigma Cat. no. E7889)
- Nuclease Free Water (Sigma cat. no. W4502)
Procedure
RNAse Free Microinjection Buffer (10 mM Tris-HCl, pH 7.4, 0.25 mM EDTA)
For 10 ml, mix together:
- 9.9 ml water
- 0.1 ml Tris-HCl, pH 7.4
- 0.005 ml EDTA
For 20 ml, mix together:
- 19.8 ml water
- 0.2 ml Tris-HCl, pH 7.4
- 0.010 ml EDTA
Wash 0.02 µm filter with 1 ml buffer – discard wash
Filter remaining buffer through filter for use as dialysis buffer or for use as microinjection buffer.
mRNA/sgRNA/oligo/plasmid Donor Preparation for Microinjection
The Transgenic Core has used Cas9, TALENs, and zinc finger nucleases to target more then 30 genes in mice and rats. The majority of projects have been gene disruptions caused by non-homologous endjoining. Eight projects used donors to introduce genetic changes by homologous recombination. The suggestions below are based our successful experience.
- mRNA and sgRNA are transcribed in vitro with kits and procedures as described (Geurts et al, 2009, Wefers et al., 2013, Yang et al., 2013). Nuclease reagents typically clog glass needles used to microinject reagents into fertilized eggs. Clogged needles lead to decreased egg survival and lower yields of genetically modified mice or rats. To reduce clogging and improve outcomes, the Transgenic Core asks that wash buffers and elution buffers used in mRNA and sgRNA purification be pre-filtered through 0.02 um filters. Do not pass nucleic acid solutions through the filters. The pore size on the filters is small enough to trap nucleic acids. Alternatively Cas9 mRNA or Cas9 protein solutions may be purchased from commercial vendors. Whether Cas9 reagents are prepared by the submitting laboratory or obtained from a third party, the Transgenic Core recommends the use of quality control assays to demonstrate activity (i.e. in vitro translation of mRNA into Cas9 protein, Western blotting of translation product, and Cas9 protein detection with antibodies).
- Plasmid DNA donors should be purified with endotoxin free kits. Pre-filter wash buffers and elution buffers with through 0.02 um filters. Do not pass nucleic acid solutions through the filters. The pore size on the filters is small enough to trap nucleic acids. Kits that are based on columns often produce particulate contaminated DNA as shown by Montigny et al. (2003). We have observed reduced clogging when DNA is purified plasmid or BAC DNA with methods that don’t rely on columns for plasmid DNA purification (such as the Epicentre BACMAX kit).
- Oligonucleotide donors are subjected to spot dialysis to remove embryotoxic chemicals. Follow standard guidelines for working with RNA to protect against later RNA degradation.
- Fill a 10cm or 15cm Petri dish with nuclease free microinjection buffer. Place a Millipore dialysis filter on the surface of the buffer so that it floats (place the filter shiny side up).
- Carefully spot the nucleic acid solution into the center of the filter. Replace the Petri dish lid. Dialyze for 30-60 minutes. Up to 200 ul can be placed on a filter without losing it to the buffer. Leave the dialysis to proceed quietly without any shaking or movement. Do not let the dialysis to go more than 3 hours; otherwise the drop might begin to evaporate.
- Carefully Pipette off the solution. Place the tip in the middle of the droplet and carefully aspirate as much as possible without stopping. Transfer the nucleic acid solution to a sterile microtube. Quantitate and store at – 80°C. Recoveries between 50-70% of the original volume are normal. The rest remains attached as a very thin liquid layer onto the surface of the filter and is difficult to pipette it off.
- The Transgenic Core microinjects nuclease reagents into the pronucleus. This has been demonstrated to be superior to cytoplasmic injection for protein expression from mRNA (Wefers et al., 2013). The efficiency of pronuclear microinjection and cytoplasmic microinjection is the same (for example the data in Table S1 of Yang et al. shows that for every 100 embryos undergoing pronuclear microinjection with 5 ng/ul Cas9 mRNA + 2.5 ng/ul sgRNA + 10 ng/ul Oct4-GFP donor DNA that 13.3 genetically modified embryos were produced. For every 100 embryos undergoing cytoplasmic injection with 100 ng/ul Cas9 mRNA + 50 ng/ul sgRNA + 200 ng/ul Oct4-GFP donor DNA 13.6 genetically modified embryos were produced.)
- Mix together nucleic acids in 0.02 um filtered RNAse Free Microinjection Buffer at the desired concentrations. Prepare fifteen aliquots of 50 ul in 1.5 ml microtubes. Store at -80°C.
| Suggested Concentrations | ||
Cas9 mRNA 5.0 ng/µl | sgRNA 2.5 ng/µl | Oligonucleotide or Plasmid Donor 10 ng/µl |
| 5’ TALEN 2.5 ng/µl | 3’ TALEN 2.5 ng/µl | Oligonucleotide or Plasmid Donor 10 ng/µl |
| 5’ ZFN mRNA 2.5 ng/µl | 3’ ZFN mRNA 2.5 ng/µl | Oligonucleotide or Plasmid Donor 10 ng/µl |
Bibliography
Geurts AM, Cost GJ, Remy S, Cui X, Tesson L, Usal C, Menoret S, Jacob HJ, Anegon I, Buelow R. 2009. Generation of Gene-Specific Mutated Rats Using Zinc-Finger Nucleases. Methods in Molecular Biology. 597: 211- 225.
Montigny WJ, Phelps SF, Illenye S, Heintz NH. 2003. Parameters influencing high-efficiency transfection of bacterial artificial chromosomes into cultured mammalian cells. Biotechniques. 35:796-807.
Wefers B, Panda SK, Ortiz O, Brandl C, Hensler S, Hansen J, Wurst W, Kühn R. 2013. Generation of targeted mouse mutants by embryo microinjection of TALEN mRNA. Nat Protoc. 8:2355-2379.
Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. 2013. One-Step Generation of Mice Carrying Reporter and Conditional Alleles by CRISPR/Cas-Mediated Genome Engineering. Cell. 154:1370-1379.
Activity of CRISPR/Cas9
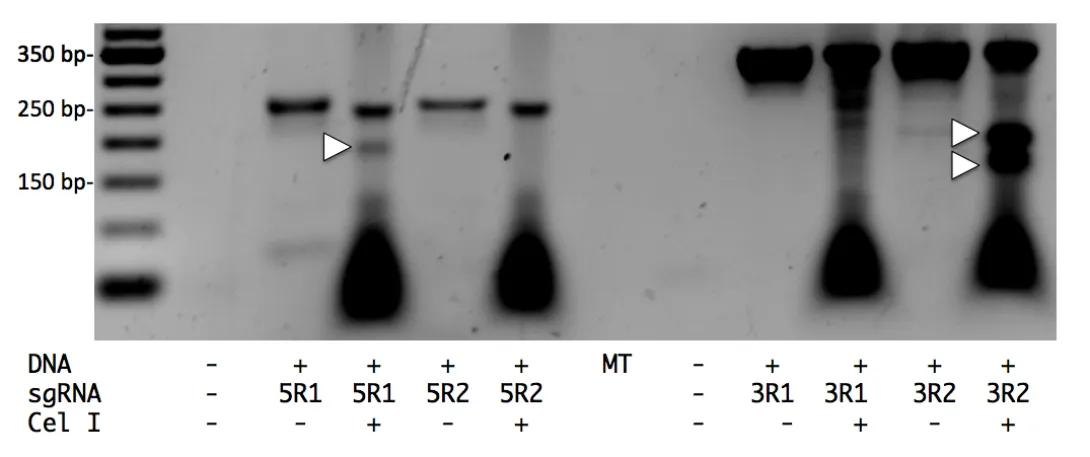
DNA: + indicates genomic DNA from JM8A3 ES cells transfected with 15 ug PNm pX330 plasmid and 5 ug PGKpuro plasmid placed under 2ug/ml puromycin selection for two days, and cultured another four days before DNA extraction.
5R1 and 5R2: pX330 plasmids targeted to 5’ side of exon.
3R1 and 3R2: pX330 plasmids targeted to 3’ side of exon.
Cel I: + indicates addi*on of Cel I enzyme to PCR product.
MT: empty lane.
Arrowheads indicate DNA fragments produced by Cel I cleavage aXer mutant and wild type DNA strands present in PCR product were denatured and annealed.
Activity of Endonucleases
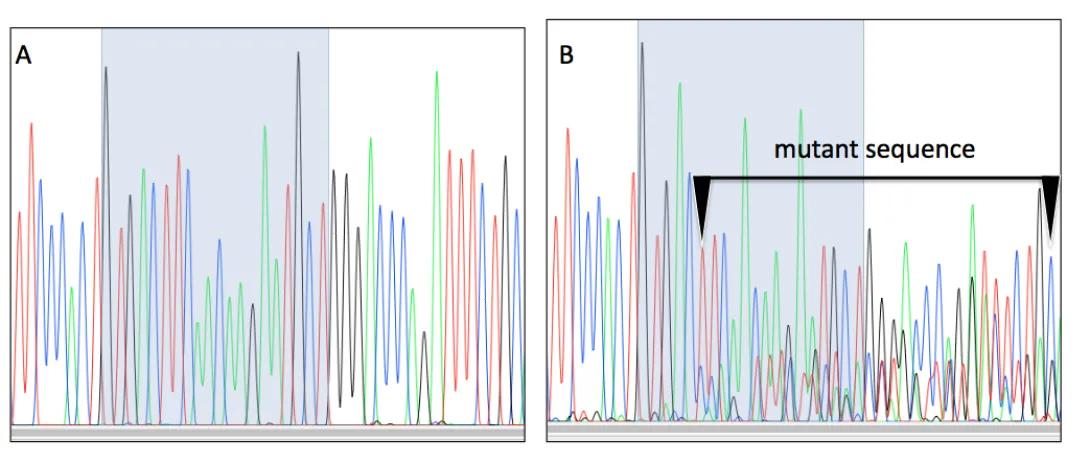
Fertilized mouse eggs were microinjected with endonucleases to cut genomic DNA. Eggs were cultured to the blastocyst stage. PCR was used amplify a DNA surrounding the cut site. The PCR product was purified from an agarose gel and submitted for DNA sequencing. Blue shading indicates sgRNA sequence.
Panel A: DNA sequencing chromatogram showing only wild type genomic DNA is present in the blastocyst.
Panel B: DNA sequencing chromatogram showing multiple DNA sequences are present in the blastocyst. This indicates that nuclease cut the chromosome and the egg repaired the damage by non-homologous endjoining.
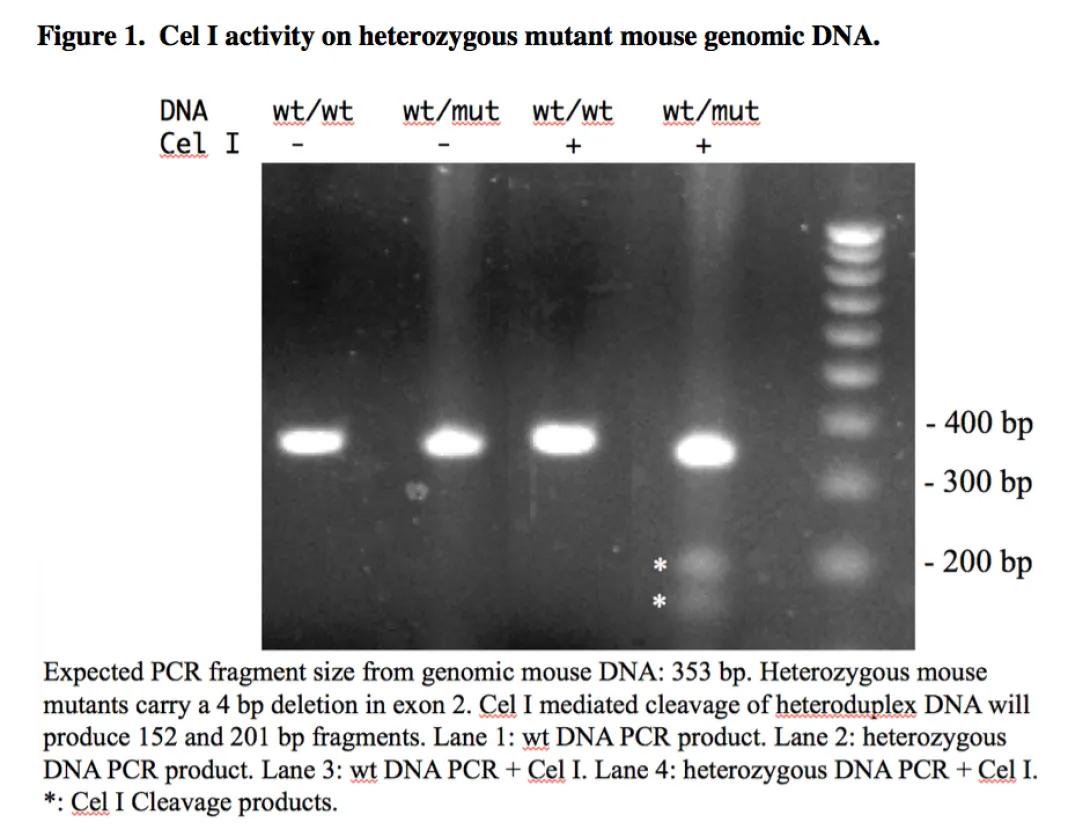
Protocol: Cel I Nuclease Assay
Materials
- PCR reaction tube of potentially heterozygous DNA sample
- PCR tubes, Pipetman, and tips
- Crude Cel I (celery extract)
- Microwave Oven
- Glass beaker
- Microcentrifuge tube floating rack
- PCR thermal cycler or other heat block set to 45C
- Stop buffer: mix one volume of loading dye with one volume of 500 mM EDTA
- 1.5% Agarose gel in TBE
Procedure
- Denature PCR amplified DNA:
- Place 4 ul of PCR reaction in a PCR tube
- Heat 200 ml of water in a glass beaker in a microwave oven to boiling Remove beaker from microwave oven
- Immediately add PCR tube(s) in floating rack to hot water
- Wait until water cools to room temperature
- Cel I Treatment:
- Remove PCR tube from floating rack
- Spin down tube in microcentrifuge
- Add 6 ul of crude Cel I
- Incubate at 45C for five minutes
- Place tube on ice
- Add 5 ul stop buffer
- Place tube on ice
- Gel Analysis:
- Load samples onto 1.5% agarose gel
- Apply 50 V and slowly run the sample into the gel
- Photograph gel and store image in archive
Reference:
Otto EA, Helou J, Allen SJ, O’Toole JF, Wise EL, Ashraf S, Attanasio M, Zhou W, Wolf MT, Hildebrandt F. 2008. Mutation analysis in nephronophthisis using a combined approach of homozygosity mapping, CEL I endonuclease cleavage, and direct sequencing. Hum Mutat. 29:418-26.
Celery Juice Extract Protocol
Kindly Provided by Susan Allen ([email protected])
- Rinse and dry celery stalks. Trim away leaves and bottom white parts. Weigh trimmed celery (Actual weight_____gm)
- In cold room set up juicer. Juice celery. (Actual volume _____ ml)
- In cold room set up stirrer with beaker of celery juice. Adjust to FINAL conc of 0.1 M Tris HCl, pH 7.7, 100 μm PMSF
Initial vol of CJ = _____ml + Stock Tris vol _____ml + Stock PMSF vol ____ ml = Total ________ml
Stock Tris = 1M Tris HCl, pH 7.7
For 1 liter of 1 M use 121.14 g then pH to 7.7 with conc HCl
Need about 4 liters for 1 bunch of celery.
Use 10 ml stock for every 100 ml Final Volume.
Stock PMSF = 0.1 M PMSF in isopropanol ( preferably anhydrous)
For 100 ml use 1.742 g then heat @ 35ºC for approx 10 min (may need longer)
Use 0.1 ml stock for every 100 ml Final Volume. - Spin (Beckman J221/ Rotor JA 14/ 2600 g or approx 4000 rpm/ 20 min/ 4ºC)
- Pellet: DISCARD, Sup: KEEP
- Bring supernatant to 25% (NH4)2SO4.In cold room with gentle stirring add 144 g of (NH4)2SO4 per liter of sup. Mix at least 30 min.
- Spin (Beckman J221/ Rotor JA 14/ 15,300 g or approx 10,000 rpm/40 min/4ºC)
- Pellet: DISCARD, Sup: KEEP
- Bring supernatant to 80 % (NH4)2SO4. In cold room with gentle stirring add 390 g of (NH4)2SO4 per liter of sup. Mix at least 30 min (or O/N).
- Spin (Beckman J221/Rotor JA 14/15,300 g or approx 10,000 rpm/ 90 min/4ºC)
- Pellet: KEEP (can store at –80ºC ), Sup: DISCARD SLOWLY
- Resuspend pellet in 0.1M Tris HCl, pH 7.7, 100 μM PMSF in final vol equal to one tenth of starting volume. Keep on ice.
Pellet volume:____________1/10th starting volume_________ - Transfer to dialysis tubing (Spectra /Por, 10,000 MWCO). Prepare tubing by soaking in H20 to remove Na Azide (change every hour – 6 changes total)
- Dialyze in cold room against 0.1M Tris HCl, pH 7.7, 100 μM PMSF. Use 7 changes of media with the first 4 changes lasting approx 1 hour each
4 liters = 400 ml of Stock Tris
4.0 ml of Stock PMSF
qs to 4 liters with H20
1 X 4L___________
2 X 4L___________
3 X 4L___________
4 X 4L___________
5 X 4L___________
6 X 4L___________
7 X 4L___________ - Buffer as below then store celery juice extract (CJE) in 10 ml aliquots @ -20ºC.
Yield:___________
For each 10 ml aliquot add to buffer: 10 μl of Triton X-100
100 μl of 1M KCl
100 μl of 1M MgCl2
100 μl of 100x BSA stock
References:
Till, BJ et al Nuclei Acids Research (2004) Vol 32 #8, p. 2632-2641
Till, BJ et al Methods in Molecular Biology (2003) Vol 236, p. 205-220
Materials
- One 10cm gelatin coated dish of JM8.A3 ES cells in exponential growth (renew in the morning if you will do the electroporation in the afternoon)
- Electroporation medium
- KO-BL6 ES Cell Culture medium
- Puromycin Selection medium
- Trypsin + Chicken serum (T+CS)
- D-PBS
- 2 gelatin-coated 10cm dishes
- Electroporation cuvette, 0.4 cm gap (BioRad # 165-2088)
(See below for medium protocols)
Plasmids in TE:
1. PGKpuro
2. pX330 (Cas9) with desired sgRNA
construct name:_________conc:_____μg/μl, μl for 15 μg____
OR
3. X461(Cas9n) + desired sgRNA #1
construct name:___________conc:_____μg/μl, μl for 10 μg____
pX461(Cas9n) + desired sgRNA #2
construct name:___________conc:_____μg/μl, μl for 10 μg____
Method
- Harvest JM8.A3 ES cells from plate by trypsinization with T+CS. Resuspend at a concentration of 107 cells/ml in electroporation medium.
- Add DNA and cells to electroporation cuvette.
- Add 15 ìg X330 plasmid (Cas9) with desired sgRNA in TE, or 10 μg each of X461 plasmids (Cas9n) with desired sgRNAs.
- Add 5 μg PGK-puro.
- Add 0.8 ml of cells at 107cells/ml.
- Pipet gently up and down without introducing air bubbles into the sample.
- Electroporate with BioRad electroporator at room temperature. 250 μF and 0.3 Kev.
- Time constant = _______
- Dilute cells in 20 mls KO-BL6 media + penstrep.
- Dispense cells to 2 x gelatin-coated P100s. Gently slide dishes forth and back, then side to side, to distribute the cells evenly across the surface.
- Renew next day with KO-BL6 media + penstrep.
- For next two days, renew daily with KO-BL6 media + penstrep + 2 μg/ml puromycin.
- For next four days, renew daily with KO-BL6 media + penstrep.
- If the colonies are very small, or you don’t see any, let the dishes remain in the incubator for 2-4 more days without changing the media.
- Harvest cells from P100s by trypsinization, pooling both duplicate P100s. Wash cell pellet 2x in D-PBS. Resuspend final cell pellet in 0.2 ml PBS and transfer to microcentrifuge tube for DNA extraction by Qiagen DNeasy kit.
Trypsin + Chicken Serum
- Add 5 ml chicken serum, 5 ml 2.5% trypsin solution, and 186 mg EDTA to 475 ml PBS.
- Sterile filter, aliquot and store aliquots at -20°C.
- PBS (ThermoFisher Catalog no. 14190094)
- Chicken Serum (ThermoFisher Catalog no. 16110082)
- 2.5% trypsin solution (ThermoFisher Catalog no. 15090046)
- EDTA powder (Sigma-Aldrich Catalog no. E6758)
Electroporation Medium
- High Glucose DMEM (ThermoFisher catalog no. 11965092)
- 4mM Glutamine (ThermoFisher catalog no. 25030081)
- 0.1 mM 2-mercaptoethanol (Sigma-Aldrich catalog no. M-7522)
- Penicillin/Streptomycin 50 units/ml (ThermoFisher catalog no. 15070063)
- 15% ES cell qualified fetal bovine serum
- 1000 U/ml LIF (EMD Millipore catalog no. ESG1107)
KO-BL6 ES Cell Culture Medium
- KO-DMEM (Invitrogens 10829
- 4mM Glutamine (ThermoFisher catalog no. 25030081)
- 0.1 mM 2-mercaptoethanol (Sigma-Aldrich catalog no. M-7522)
- Penicillin/Streptomycin 50 units/ml (ThermoFisher catalog no. 15070063)
- 15% ES cell qualified fetal bovine serum
- 1000 U/ml LIF (EMD Millipore catalog no. ESG1107)
- 1% Non-essential amino acids (ThermoFisher catalog # 11140050)
Selection Medium
- Add Puromycin to KO-BL6 ES Cell Culture Medium
- 2 μg/ml puromycin (ThermoFisher catalog no. A1113803)
For additional information on how to culture mouse ES cells see: Hughes ED, Saunders TL. 2011. “Gene Targeting in Embryonic Stem Cells” in Advanced Protocols for Animal Transgenesis: An ISTT Manual. S Pease and TL Saunders (eds) Springer-Verlag, Berlin. pp. 291-325.
"Hotshot" Genomic DNA Preparation
(Hot sodium hydroxide and tris)
From Biotechniques. 2000 Jul;29(1):52,54
Alkaline Lysis Reagent
| Reagent | [Final] | Add | Of |
| NaOH | 25mM | 125λ | 10N NaOH |
| EDTA | 0.2mM | 20λ | 0.5M EDTA |
| 50ml | ddH2O |
pH will be 12
EDTA = disodium EDTA
Neutralization Buffer
| Reagent | [Final] | Add | Of |
| Tris-HCl | 40mM | 325mg | Tris-HCl |
| 50ml | ddH2O |
pH will be 5
Protocol
- Obtain tissue
- 0.2 cm tail snip
- 2 mm ear biopsy
- Place tissue in 96-well plate
- Add 75λ of Alkaline Lysis Reagent
- Heat to 95℃ for 10 min to 1 hr (30 min is optimal)
- Cool to 4℃
- Add 75λ Neutralization Buffer
- Use 1 to 5λ per PCR reaction
Notes
- DNA is suitable for PCR reactions but NOT for Southerns
- Heating for longer than 30 minutes does not increase [DNA]
- pH of reagents does not need to be altered
- Do not worry about undigested floating tissue
- DNA yield is similar for tail snips and ear punches
- Too much tissue will destroy PCT attempts
- DNA must be stored at 4C or -20C
From Mike Charles 10/15/03
[email protected]
Beta Galactosidase (lacZ) Staining
This procedure describes how to process samples for lacZ staining. Beta galactosidase is an enzyme that hydrolizes beta galactosides. The cleavage of Xgal (5-bromo-4-chloro-3-indolyl-b -galactopyranoside) results in a dark blue precipitate. A nuclear localized lacZ transgene can be used to mark transgene expressing cells unambiguously (endogenous enzyme activity is cytosolic). If desired, antibody staining can be carried out for cytosolic proteins (see Brinkmeier et al. ). Thick specimens, such as late stage mouse embryos, can be cleared by treatment with (see Turkay et al.). Background staining can be reduced by increasing the pH of the phosphate buffers used to process samples. For a review see the chapter by Saunders.
Saunders TL. 2003. Reporter molecules in genetically engineered mice. Methods Mol Biol. 209:125-143.
Turkay, A, Saunders, T, Kurachi, K. 1999. A method for histochemical analysis of cleared mouse embryos and adult tissues expressing ß-galactosidase. J. Histotechnology. 22:323-324.
lacZ Staining Procedure for Cells and Mouse Embryos
- Rinse cells (or mouse embryos) once in phosphate buffer pH 7.3 at room temperature.
- Fix cells for 5 minutes (embryos for 15-30 minutes, depending on size) at room temp.
- Wash cells 3 times for 5 minutes (15 minutes for embryos) at room temperature.
- Stain cells (embryos) 4 hours to overnight at 37*C depending on level of LacZ activity.
- After staining, pour off stain, replace with wash buffer.
- Store samples at 4*C. Staining will intensify in wash buffer at 4*C.
Notes on Staining
The procedure doesn’t have background problems in most cell types. With embryos, background staining is seen in the yolk sac of day 10 embryos and on. In addition, a thin stripe of staining is observed in the hindbrain of day 12 embryos. If background is a problem, then try increasing the pH of the phosphate buffer. Staining procedure works at pH 8.5.
Staining Procedure for Cryostat Sections
- Collect tissues or embryos and place in isopentane at -30 to -45*C for 30 seconds.
- Store frozen samples at -70*C.
- Section embryo.
- Fix sections for 5 minutes- use glutaraldehyde fix.
- Wash 3 times for 5 minutes.
- Stain sections overnight at 37*C.
- After staining, pour off and save stain, replace with wash buffer. Can counterstain with neutral red, dehydrate in ethanol and xylene and coverslip.
- Store samples at 4*C. Staining will intensify in wash buffer at 4*C
Materials and Reagents
0.1 M phosphate buffer, pH 7.3
115 ml 0.1 M sodium phosphate, monobasic (6.9 g in 500 ml H2O)
385 ml 0.1 M sodium phosphate, dibasic (14.2 g in 500 ml H2O)
500 ml total
Fix Solution – Always prepare fresh
4 ml 25% glutaraldehyde
2.5 ml 100 mM EGTA, pH 7.3
0.4 ml 1 M MgCl2
173.1 ml double distilled H2O
20.0 ml 10x PBS
200.0 ml total
Wash Buffer
0.4 ml 1 M MgCl2
2.0 ml 1% deoxycholate (may be omitted)
2.0 ml 2% NP-40
195.6 ml 0.1 M sodium phosphate, pH 7.3
200.0 ml total
X-gal stock 250 mg X-gal
(5-bromo-4-chloro-3-indolyl-b -galactopyranoside) (Sigma B4252)
10 ml dimethyl formamid (Sigma D4551)
X-gal stain – Make fresh
2.0 ml 25 mg/ml X-gal stock
0.106 g potassium ferrocyanide (Sigma P-9387)
0.082 g potassium ferricyanide (Sigma P-8131)
48.0 ml wash buffer
50.0 ml total
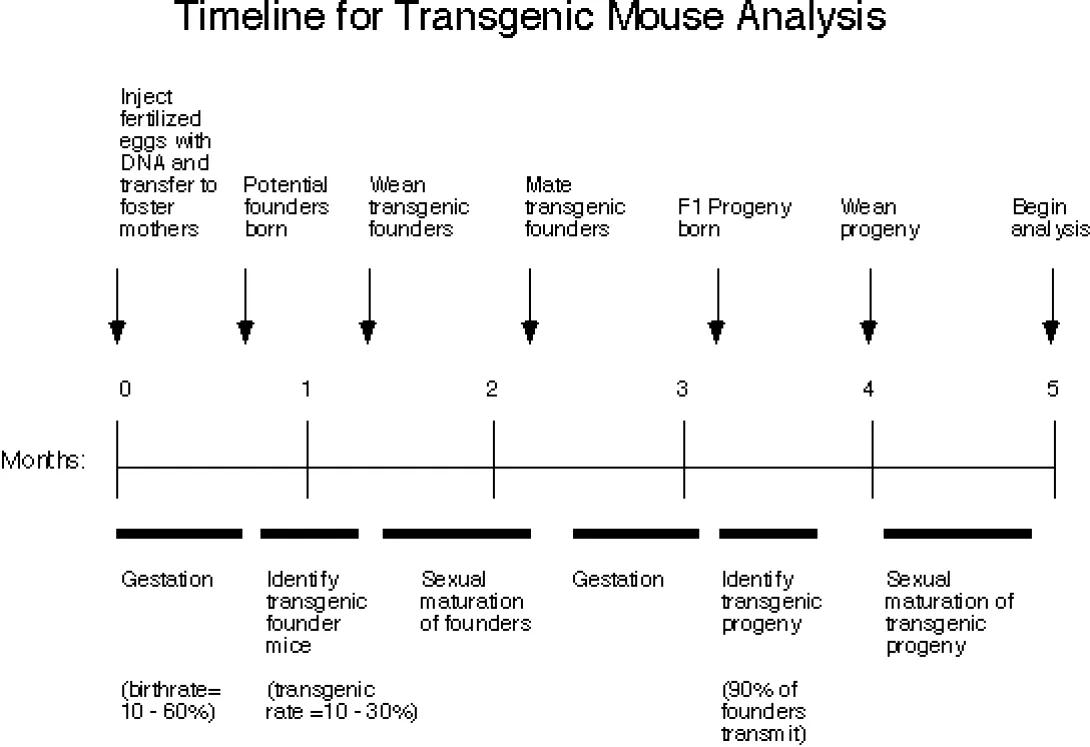
Also see the Transgenic Mouse & Transgenic Rat Outline
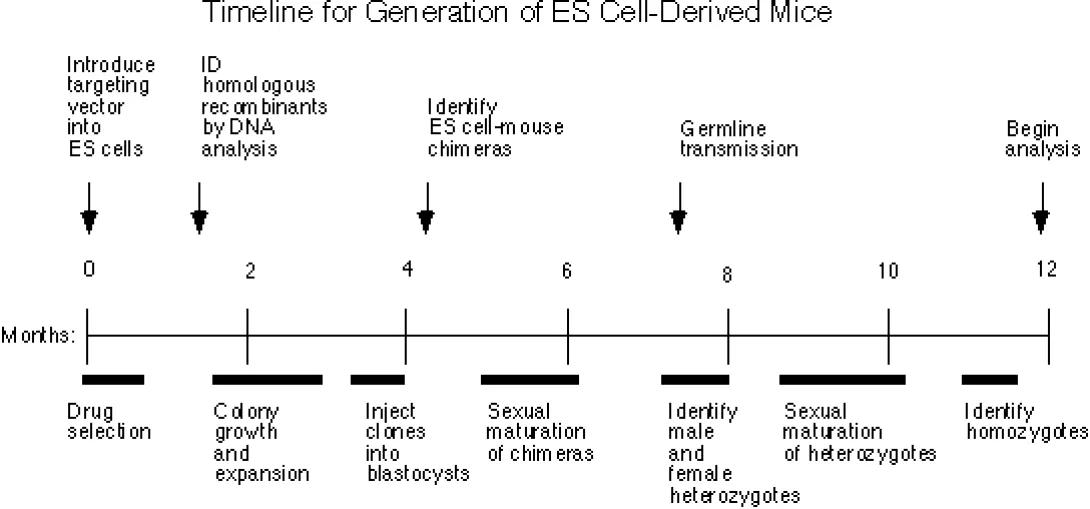
Also see the Transgenic Mouse & Transgenic Rat Outline
Chromosome Counts for ES Cells – Testing for Euploid Clones
- Why count chromosomes in gene targeted ES cells?
It is recognized that not all cell lines in culture have normal chromosome complements. Numerous groups have observed that aneuploid ES cell clones do not form germline ES cell-mouse chimeras (exception: germline transmission from X:O ES cells can occur through female chimeras). We routinely detect aneuploid ES cell clones in our experiments, in gene trap clones imported from international consortia, and in clones generated independently by other laboratories. Deriving and breeding ES cell chimeras from aneuploid ES cells is expensive with respect to animals used, time, and money. The simple chromosome counting method below can improve the efficiency of germline transmission in any experiment. We count 20 spreads per ES clone in our Transgenic Core Facility. We microinject ES cell clones in which 60% or more of the spreads contain 40 chromosomes. Note that simply because most of the chromosome spreads in an ES cell clone are euploid is no guarantee that the clone will go germline. - Why not karyotype chromosomes in gene targeted ES cells?
A karyotype shows and identifies each chromosome in a spread. This identification of the chromosomes in multiple spreads is a time consuming process. Commercial services for karyotyping are available, but can add dramatically to the cost of a gene targeting project. Although some ES cell lines with 40 chromosomes will be aneuploid because of chromosome duplications and deletions, the proportion of ES cell clones with euploid chromosome counts that go germline is so high that there is no real advantage to screening the cells by karyotype analysis.
View Chromosome Counting Protocol
Genotyping Protocols
PCR Analysis of Tail DNA
This is how we test mice and rats for the presence of the transgene by PCR. It is provided for those investigators who are unfamiliar with DNA genotyping and who need to establish a genotyping method.
- Quantitate tail DNA.
- Prepare 20 µl aliquots of tail DNA diluted to 200 ng/microliter in autoclaved water.
- Heat DNA for 1 minute in a dry bath set for 95 degrees C. Spin down tubes place on ice.
Prepare PCR cocktail.
Reagents for 1 reaction for 20 reactions autoclaved water 14.0 microliters 280.0 microliters 10X PCR Buffer 2.5 microliters 50.0 microliters Nucleotides (10 millimolar) 4.0 microliters 80.0 microliters Primer A (10pmol/microliter) 1.25 microliters 25.0 microliters Primer B (10pmol/microliter) 1.25 microliters 25.0 microliters Taq Polymerase (0.3125 U/microliter) 1.0 microliters 20.0 microliters Total Volume 24. microliters 480.0 microliters - Aliquot 24.0 microliter of cocktail into PCR tubes.
- Add 1.0 microliter of heat treated DNA to PCR tube (if tail DNA is less than 200 ng/microliter then add 200 ng of DNA in a volume up to 3 microliters).
- Final volume of PCR reaction is 25.0 microliters.
- Overlay with mineral oil and place into thermal cycler.
- Amplify 30 to 35 cycles.
- Analyze on appropriate ethidium bromide containing agarose gel. We analyze up to 60 samples on a 20 X 20 cm 1.5% agarose gel in TBE.
Tips
- Prepare a cocktail with an extra 10% to account for pipetting errors.
- Remember to include a negative control of water only and a positive control of known transgenic mouse DNA or linearized plasmid DNA diluted in mouse tail DNA to the one copy level.
- Remember to include an internal positive control to ensure that the DNA sample is “amplifiable.” For this purpose we use primers for mouse beta globin. Primers that amplify any single copy mouse gene may also be used. For example, amplification of beta globin indicates that the DNA is clean and “amplifiable.” This will prevent false negative results and positive animals will not be accidentally discarded.
- Internal positive control primers may be combined with transgene specific primers if the two primer sets do not interfere with each other.
- Use primer picking software. We have had very good results with Primer3.
Preparation of Copy Standards for PCR Genotyping Sensitivity and Southern Blot Copy Number Determination
PCR screens must be designed to detect transgene DNA at the single copy level or 0.1 copy level..
Southern Blots analysis of transgenic mice need copy standards to estimate copy number.
Copy standards are prepared by mixing non-transgenic tail DNA with a known amount of transgene DNA is to produce transgene copy standards.
For PCR, these standards can be used to determine the sensitivity of the PCR assay. You must obtain single copy sensitivity in your PCR prior to transgene submission. When you test DNA from potentially transgenic founder mice, run your single copy PCR test sample to ensure that your PCR assay is sensitive enough to avoid false negatives.
Southern Blots are commonly used to determine transgene copy number and the number of integration sites in transgenic founder mice. Download a pdf file illustrating Southern Blot analysis of transgenic founders.
Calculation of copy number standards
Assumption: the Haploid content of a mammalian genome is 3 X 109 bp
Assumption: you have 2 micrograms of tail DNA available
Since the transgenic founder mice are hemizygous:
mass of transgene DNA = N bp transgene DNA
1 microgram genomic DNA 3 X 109 bp genomic DNA
Example: for a 5,480 bp transgene insert or plasmid
mass of transgene DNA = 5,480 bp cloned DNA or
1 micrograms genomic DNA 3 X 109 bp genomic DNA
mass of transgene DNA = (5,480 bp cloned DNA) X (1 µg genomic DNA) or
3 X 109 bp genomic DNA
mass of transgene DNA = 1.83 picograms per 1 ug genomic DNA
transgenic mice will be hemizygous for the transgene, not homozygous
1.83pg transgene should be added to 2 ug of genomic DNA
Thus, to prepare a 1 copy standard: add 1.83 pg of transgene DNA to 2 microgram tail DNA
0.1 copy 0.183 pg
10 copy 18.3 pg
50 copy 91.5 pg
100 copy 183 pg
For use as a transgene PCR standard, use 200 ng of the spiked tail DNA as a substrate in a 25 ul PCR reaction as described: genotyping transgenic mice.
For use in Southern blot analysis, digest the tail DNA as you would for Southern analysis, and add the transgene insert DNA (not the entire plasmid) just before you load your gel. Remember to reserve one lane for genomic DNA only with no spike. For an example of copy standards in Southern blots, refer to Camper SA. 1987. Research applications of transgenic mice. Biotechniques 5, 638-650.
Click here for more review articles.
This primer pair is used to genotype transgenic mice that carry the beta galactosidase (lacZ) reporter gene. The primers recognize sequences within the beta galactosidase gene.
5' TTC ACT GGC CGT CGT TTT ACA ACG TCG TGA 3' 30mer
5' ATG TGA GCG AGT AAC AAC CCG TCG GAT TCT 3' 30mer
Size of expected amplification product is 364 bp.
Thermal cycler profile:
94°C 60 sec
72°CC 120 sec
30 cycles
72°C 10 min
4°C Hold
This primer pair is used to amplify the endogenous rat prolactin gene. Amplification will thus demonstrate that the DNA sample is a good substrate for PCR.
5′ GCT TCT GAG CAA TGA CAC CA 3′ 18 mer
5′ ATT CCA GGA GTG CAC CAA AC 3′ 18 mer
Size of expected amplification product is 391 base pairs.
Thermal cycler profile:
- 95°C for 45 seconds
- 95°C for 45 seconds
- 55°C for 1 minute
- 72°C for 30 seconds
- Go To #2 30 times
- 72°C for 10 minutes
- Hold at 4°C
Rat Prolactin Sequence Source (Genbank Accession Number AH002235):
Gubbins EJ, Maurer RA, Lagrimini M, Erwin CR, Donelson JE. Structure of the rat prolactin gene. J Biol Chem. 1980 Sep 25;255(18):8655-62.
Isolation of DNA from Mouse Tail Biopsies
- Obtain tail biopsies from 2 to 3 week old mice: Hold mouse firmly at base of tail with one hand, with the other cut off 0.5 to 1.0 cm of the tail tip with a scalpel or single edge razor blade.
- Add 600 microliters of TNES and 35 microliters Proteinase K (10 mg/ml).
- Incubate overnight (8-24 hr) at 55°C.
- Add 166.7 microliters 6M NaCl. Shake vigorously for 15 sec.
- Microfuge (12,000-14,000 xg) for 5 min at room temp.
- Remove supernatant to new tube and add 2 volumes cold 95% ethanol (EtOH).
- Spool precipitated DNA with closed end capillary tube.
- Rinse DNA pellet with 70% EtOH (dipping in a tube filled with 70% EtOH works well) and allow to air dry 5-10 min.
- Resuspend in 100-500 microliters of TE (10mM Tris, pH 8.0, 1mM EDTA). Volume depends empirically on pellet size.
- Heat at 65°C for 10 minutes to aid dissolution of DNA.
- Quantitate DNA and store at 4°C until needed.
TNES
10 mM Tris, pH 7.5
400 mM NaCl
100 mM EDTA
0.6% SDS
“6M” NaCl
Saturated salt solution stored at 37°C
Reference:
Miller SA. Dykes DD. Polesky HF. 1988. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research. 16(3):1215.
This primer pair is used to amplify the endogenous mouse beta-globin gene. Amplification will thus demonstrate that the DNA sample is a good substrate for PCR .
5′ CCA ATC TGC TCA CAC AGG ATA GAG AGG GCA GG 3′ 32 mer
5′ CCT TGA GGC TGT CCA AGT GAT TCA GGC CAT CG 3′ 32 mer
Size of expected amplification product is 494 base pairs.
Thermal cycler profile:
94°C 30 seconds
60°C 90 seconds
72°C 120 seconds
35 cycles
72°C 10 min
4°C Hold
Beta-Globin Sequence Source (Genbank Accession Number J00413):
Konkel DA, Tilghman SM, Leder P. 1978. The Sequence of the Chromosomal Mouse beta-globin Major Gene: Homologies in Capping, Splicing and Poly(A) Sites. Cell 15:1125-1132.
This primer pair is used test DNA for the presence of the neomycin gene.
The primers recognize sequences within the neor cassette of gene targeting vectors.
5′ AGG ATC TCC TGT CAT CTC ACC TTG CTC CTG 3′ 30mer
5′ AAG AAC TCG TCA AGA AGG CGA TAG AAG GCG 3′ 30mer
Size of expected amplification product is 492 base pairs.
Thermal cycler profile:
94°C 30 sec
72°C 120 sec
30 cycles
72°C 10 min
4°C Hold
Mouse Breeding
We hope that this information will be useful to investigators who are unfamiliar with mouse breeding or who are breeding transgenic or gene targeted mice for the first time. These suggestions are based on our experience. They are open to modification and should not be construed as a comprehensive set of rules.
- Keep accurate breeding records. Make a pedigree for each transgenic founder or embryonic stem cell-mouse chimera.
- Mate mice when they are sexually mature (6 to 8 weeks old). We recommend that transgenic founders or chimeras be mated to C57BL/6 mice. After 6 generations of mating to C57BL/6, more than 99% of the genetic background will be C57BL/6. By analyzing gene expression on a C57BL/6 background, any influence of the genetic background on gene expression will be controlled for by comparison to normal C57BL/6 mice. Alternatively, chimeras can be mated with 129/Sv+Tyr-c+p mice, which have the same genotype as the embryonic stem cells. This will produce mice with the targeted gene mutation on the 129/Sv+Tyr-c+p background for comparison with the mutation on the C57BL/6 background.
- Expect litters within a month of mating since female mice go into estrus every 3 or 4 days and the gestation time of mice is 19-21 days. If no litters are produced after one month you should replace the mice that you are mating with your founder. It is possible that the transgenic founder may be infertile due to consequences of transgene expression or unknown reasons. It is possible that phenotypically male chimeras may be infertile because they are the result of colonization of a female embryo by male embryonic stem cells.
- Reasons to use C57BL/6 mice.
- a) C57BL/6 is a standard inbred strain, commonly used in transgenic breeding
- b) mate 6 – 8 week-old mice for best reproductive performance
- i. replace males when they are 1 year old
- ii. replace females after 6 litters or when they are 6 months old
- c) mate a founder male with 2 females to get 2 litters in close succession
- d) mate a founder female with 1 male
- e) mice usually mate again on the day the female gives birth, resulting in a second litter 3 weeks after the first.
- f) to rapidly produce animals, rotate 2 females through a male’s cage every 1- 2 weeks
- g) house pregnant females 1 or 2 per cage to prevent crowded cages
- Common problems and solutions:
- a) female may not care for first litter add proven breeder female to cage as helper and try again
- b) female doesn’t care for any litter most often seen with 129 females kept on bedding which precludes the construction elaborate “subterranean” nests 129 mating cages should include nesting material at all times
- b) male may cannibalize litter remove male from mating cage before female gives birth
- c) fighting mice
- i. separate the fighting mice, house them 1 per cage if necessary
- ii. females do not typically fight
- iii. males may fight in the following circumstances:
- male is placed in a cage containing other male(s)
- male is separated at weaning and then reunited with male littermates
- male is weaned into a cage that contains males from another litter
- males are aggressive and may begin to fight for no apparent reason
- adult male attacks immature female when female is placed in male’s cage
- Schedule for ear tagging, tail biopsies, weaning, and mating:
- a) record births on the cage card and the pedigree
- b) ear tag the pups when they are two weeks old
- c) obtain tail biopsies as you apply ear tags
- d) isolate tail DNA and determine genotypes before pups are 21 days old
- e) record genotypes in the pedigree
- f) wean pups when they are 21 days old
- i. remove the pups from their mothers
- ii. discard un-needed non-transgenic pups
- iii. house males and females separately
- f) when mice are 6 weeks old they may be mated (see 1. above)
Based on our experience, we propose the following guidelines for chimera breeding. If you have 10 or so chimeras to take through breeding then these guidelines apply. If you only have one or two animals then breed them indefinitely until you can produce additional animals from independent ES cell clones. You may be lucky get germline transmission at low efficiency (defined as 1 agouti pup in 200 pups born). We recommend that you breed your chimeras to C57BL/6 partners. Pups produced from sperm derived from ES cells will have agouti coats, half of these animals should carry the targeted gene. Pups that are produced from sperm derived from the C57BL/6 host embryo will have black coats.
- Discard female chimeras unless you have high contribution female chimeras and a distorted sex ratio favoring females (many female chimeras and few male chimeras). You probably injected a X:O cell line. The females may very well give germline transmission.
- Breed as many males as you can, we have observed germline transmission from males with small quantities of agouti fur.
- Breed males aggressively from the beginning ? rotate 2 females through the male’s cage every two weeks. This will require 3 cages of 2 females in addition to the male’s breeding cage. Don’t put the male chimera into a cage occupied by females, they may attack him.
- If the chimeras don’t produce pups by after 8 weeks of breeding and rotating females, they are infertile and should be discarded. If males produce 6 or more litters without transmitting then they are not likely to go germline and should be discarded.
- Establish a Pedigree for each founder animal or offspring that is mated.
- Have customized cage cards printed for mating cages and weaning cages. Mating cage cards should provide an instant summary of the activity in the cage, and serve as permanent records. The weaning card allows rapid location of mice according to line and ear tag.
- Use a customized log sheet to record ear tag number, sex, coat color, tail DNA log number, and genotype for each mouse that is tagged and tailed. Keep the log sheets with the pedigree.
- Together, the pedigree, mating cards, and log sheets should provide the following information for each mouse: birth date, sex, ear tag number, coat color, DNA log number, mating cage number, ear tag numbers of mother and father, source of mother and father, genotype, date mated, date euthanized, reason for euthanasia (e.g. tissue analysis), generation (how many times the line has been mated with C57BL/6).
Periodically we receive requests to help rescue mouse strains that are no longer breeding. Often the mice are beyond their preak reproductive years. Here are some of the approaches that can be used to “rescue” old strains.
- Use C57BL/6 F1 mice to set up new breeding cages (see mouse breeding suggestions above). Most C57BL/6 F1 hybrids have good reproductive performance. We prefer (C57BL/6J X DBA/2J)F1 animals. Outbred CD-1 and ICR mice can be used, but introduce more genetic complextiy to the line.
- Follow the breeding females for copulation plugs. Females enter estrus every 3 days or so. Mate the animals on Monday and follow them for plugs. If they haven’t plugged by Friday, keep following them over the weekend until they plug or unmate then and re-mate on Monday. Alternatively, the Transgenic Core can mate superovulated F1 female mice to your male(s) in a rederivation procedure. Eggs are collected from the females and fertilization is determined under the microscope. Fertilized eggs are transferred to pseudopregnant females to obtain pups. If the male doesn’t generate fertilized eggs (no plugs or plugs without fertilization then the next step is in vitro fertilization (see #3).
- If the F1 females plug but don’t become pregnant then the males you are using to rescue the line are subfertile, by definition. We can use euthanize the male(s), collect sperm and perform an in vitro fertilization (IVF) procedure. The Transgenic Core routinely does IVF with excellent results. Once we rescued a line from the last male even though it died two days before the IVF. In this case, the dead male was stored in a sealed plastic bag (to prevent dessication) in the refrigerator until the IVF. Another time we rescued a mouse strain from a single spontaneous male mutant with ectopic caudal limbs in the place of a penis (polypodia mutation – Ppd gene on the X chromosome).
- If the F1 males don’t plug the females then females are most likely anovulatory. In this case you can send the old females to the Jackson Laboratory for ovarian transplantation into a immunocompromised female (to prevent graft rejection) and you may be able to recover the line this way. The Transgenic Core does not offer this service.
- If you took the precaution of cryopreserving the line as frozen eggs, the Core can thaw out the eggs and recover the line. If sperm were cryopreserved we may or may not be able to recover the line by IVF or ICSI, depending on the genetic background of the sperm. We recommend that frozen sperm be tested for successful IVF before a line is discontinued.
- If the line was derived from an ES cell clone, it can be re-injected to generate a new set of germline chimeras.
- If the line is available from another laboratory you can import them.
Three transgene (Tg) transmission patterns occur in transgenic founder animals. Most founder animals transmit their Tg to 50% of their offspring. About 10% to 20% of founders are mosaic for the Tg due to late Tg integration during embryogenesis. A variable proportion of founders (5% to 30%) have more than one Tg integration site. Transmission to 30% or less of offspring is a sign that the founder’s germ cells are mosaic for the Tg. In these cases it is advisable to verify the transgenic status of the founder and to breed the mouse as efficiently as possible. Founders with more than one Tg integration may transmit the Tg to 80% or more of their offspring. Different insertion sites usually segregate independently. Southern blot analysis of these offspring is used to group mice have according to insertion site. This simplifies analysis of transgene expression since expression patterns associated each independent insertion are isolated from each other. Offspring of transgenic founders transmit the Tg as normal Mendelian gene, regardless of whether the founder was mosaic or multi-integrant.
Two transmission patterns occur in embryonic stem (ES) cell-mouse chimeras. Either they transmit or they don’t. The transmission ratios we have observed vary from 100% to 0.5%. Infertile male chimeras may also result. Since the ES cells are XY only male chimeras should be bred. We recommend that you take a male chimera through six litters before deciding to give up on it. If a chimera has a high transmission ratio, you should consider mating it directly with females from the 129 mouse strain that matches the origin of the ES cell line to obtain your targeted gene mutation on an inbred background.
We recommend that transgenic lines be maintained as hemizygotes. The primary disadvantage of maintaining lines as hemizygotes is that all offspring need to be genotyped to determine which are transgenic. Establishment of homozygous transgenic lines is is costly and entails additional breeding to produce the first homozygotes. Additional test breeding is needed to ensure homozygosity. Additional time is required to establish a homozygous line from a mosaic founder with limited transgene transmission. Extra breeding will be necessary to establish homozygous lines for each integration site in founders with multiple integrations. If this is not done carefully, animals with combinations of different integration sites will result. This will complicate interpretation of experimental results since different integration sites can have different transgene expression patterns. About 5% of transgene integration events interrupt an endogenous gene important for normal development (Meisler MH. 1992. Trends Genet. 8:341). Affected mice may display a phenotype unassociated with the transgene which may obscure the effects of transgene expression. Unequal crossing-over between transgene arrays in homozygotes may result in transgene rearrangement which will affect expression. This kind of instability occurs less often in hemizygotes. Many other difficulties can be avoided by maintaining hemizygous transgenic lines.
TABLE 1. SOME VITAL STATISTICS OF THE HOUSE MOUSE, MUS MUSCULUS
Genome
number of chromosomes 40
diploid DNA content about 6 pg per cell
3X109 bp per haploid genome
recombination units 1600 centimorgans (2000 Kb/cM)
approximate number of genes§ 0.5 – 1.0 X 105
percent of genome as five families
of highly repeated DNA sequences
(B1, B2, R, MIF-1, and EC1)¥ 8 – 10%
Reproductive Biology
gestation time 19 – 21 days
age at weaning 3 weeks
age at sexual maturity 6 – 8 weeks
approximate weight birth 1g
weaning 8 – 12g
adult 30 – 40g (male > female)
lifespan in laboratory 1.5 – 2.5 years
average litter sized 6 – 8
total number of litters per
breeding female 4 – 8
useful breeding life of females 6 – 8 months
useful breeding life of males 18 – 24 months
§McKusick, V.A. and F.H. Ruddle. 1977. The status of the gene map of the human chromosomes. Science 196:390-405.
¥Bennett, K.L., R.E. Hill, D.F. Pietras, M. Woodworth-Gutai, C. Kane-Kass, J.M. Houston, J.K. Heath, and N.D. Hastie. 1984. Most highly repeated dispersed DNA families in the mouse genome. Mol. Cell. Biol. 4: 1561-1571.
Ä Parameters such as gestation time, weight, lifespan, etc., vary between the different inbred strains.
?Litter size depends on the number of eggs liberated at ovulation, the male’s sperm count, and the rate of prenatal mortality. These may vary with age of mice, parity, and environmental conditions (e.g. diet, stress, presence of strange male) and with strain (reflecting genetic factors such as efficiency of placentation). Prenatal mortality in inbred strains can be around 10-20%.
Table 1 is adapted from Manipulating the Mouse Embryo: A Laboratory Manual, by B. Hogan, R. Beddington, F. Constantini, and E. Lacey, published by Cold Spring Harbor Laboratory. This book has an excellent bibliography for information about mouse strains and breeding.
An excellent source is the second edition of Biology of the Laboratory Mouse, by the Staff of the Jackson Laboratory, published by Blakiston Division of McGraw-Hill.
Additional information on mouse husbandry can be found in:
- Handbook on Genetically Standardized JAX Mice, 1997. by the Staff of the Jackson Laboratory, available from The Jackson Laboratory.
- Systematic Approach to Evaluation of Mouse Mutations. 2000. Sundberg, JP and Boggess D, eds. CRC Press, LLC, Boca Raton, Florida.
- Mouse Genetics and Transgenics: A Practical Approach. 2000. Jackson, IJ, and Abbott, CM, eds. Oxford University Press, New York.
- Mouse Genetics: Concepts and Applications.1995. Silver, LM. Oxford University Press, New York. (Available online).
- The NIH Guide for the Care and Use of Laboratory Animals is available online.
Some investigators will need to know the alleles at various polymorphic loci carried by the transgenic founders. This information can be found by looking up the genotypes of the mice used to obtain fertilized eggs for microinjection in:
Genetic Variants and Strains of the Laboratory Mouse, eds. M.F. Lyon and A.G. Searle, Chapter 17; “Strain Distribution of Polymorphic Variants” by T.H. Roderick and J.H. Guidi, published by the Oxford University Press.
Questions?
Contact Us
1150 West Medical Center Drive
Ann Arbor, MI 48109