Histone Modifications

Mapping genome-wide distribution patterns of specific histone modifications.
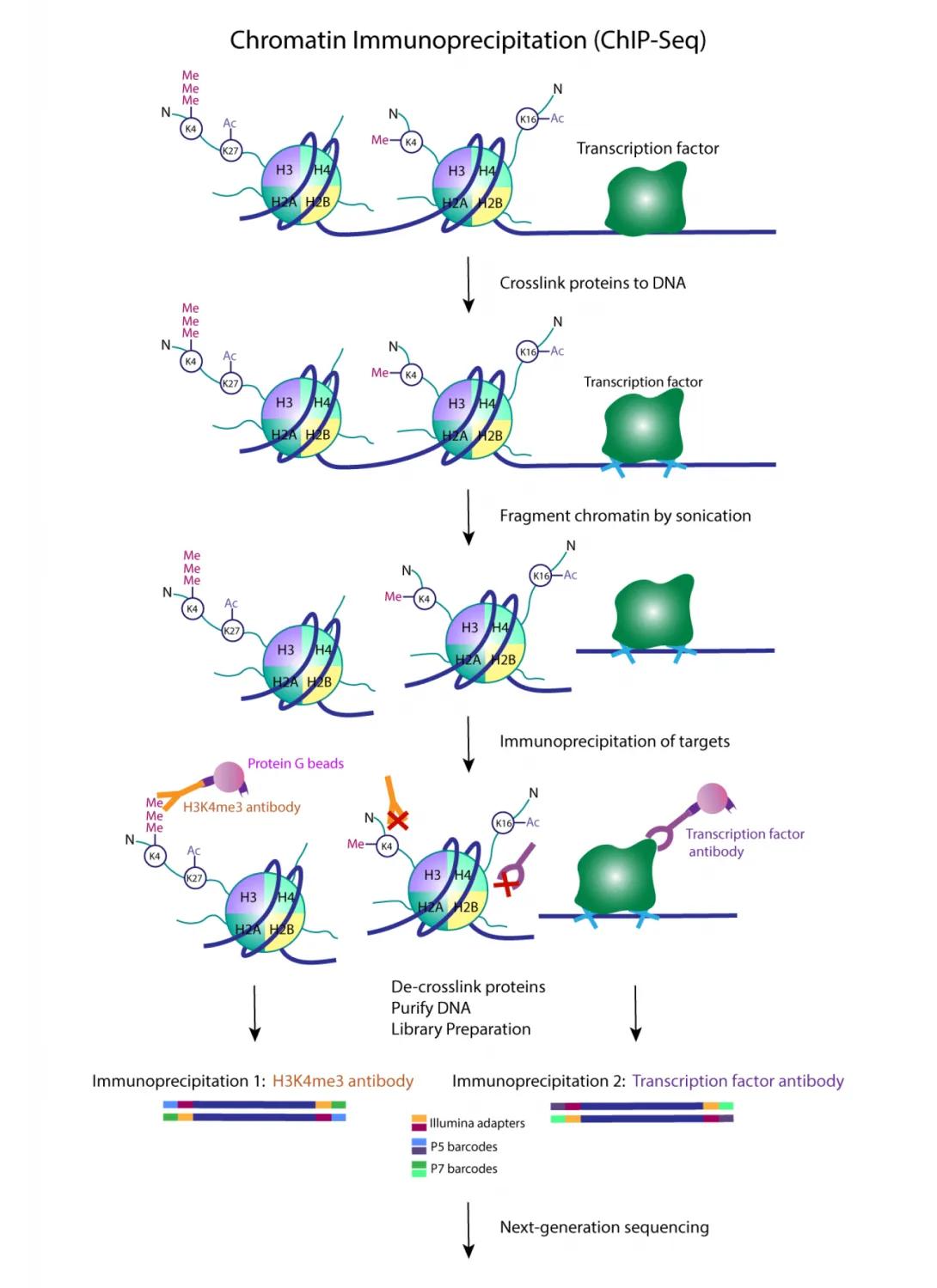
Chromatin immunoprecipitation followed by massively parallel DNA sequencing (ChIP-seq) is a method used to analyze DNA-protein interactions and epigenetic marks in a genome-wide manner at base-pair resolution. It can be used to map binding sites for transcription factors and other DNA binding proteins and to profile histone modifications associated with active or repressed transcription. Major advantages of ChIP-seq over array-based techniques (e.g. ChIP-chip) are an increase in resolution, reduction in noise/fewer artifacts, greater coverage, and a larger dynamic range.
ChIP-seq Procedure
ChIP-seq typically starts with crosslinking of DNA-protein complexes in cells. Chromatin is isolated from the cells and fragmented by sonication. ChIP-seq can also be performed on native chromatin, which is suitable for profiling histone modifications. Native chromatin is treated with MNase to fragment it into nucleosomal complexes without fixing cells. The chromatin fragments are enriched using antibodies specific to the proteins or histone modifications of interest. Crosslinks are reversed and the DNA is extracted.
The customer delivers the immunoprecipitated DNA to the Epigenomics Core for library preparation. Briefly, this entails end repair of the DNA fragments and ligation of Illumina-compatible adapters. PCR amplification incorporates indexed primers to barcode the sample and increases the amount of templates available for sequencing. Libraries are QC’d and quantified via qPCR prior to pooling and submission to the Advanced Genomics Core for next-generation sequencing.
ChIP-seq Sample Guidelines
At sample submission, the customer must provide the following to the Core. Customers submitting samples without providing the preliminary QC data specified below will be charged for library preparation. These requirements are based on the assumption that the customer wants the samples to be processed for library preparation:
- At least 5 ng of input (cross-linked and fragmented but not immunoprecipitated) and immunoprecipitated DNA for each sample. At least two biological replicates per experimental group are advised.
- Image of DNA fragmentation QC (gel image or Bioanalyzer/TapeStation results), showing the majority of fragments in the range of 200-500 bp.
- IgG immunoprecipitated samples (generating random DNA) for each experimental condition may be provided. ideally, they will only be used at the QC stage, unless they contain as much DNA as the immunoprecipitation samples.
- If available: percent input enrichment qPCR results for both positive and negative controls regions, and IgG control (as excel spreadsheet or table).
- If available: primers for positive/negative control genes to test fold enrichment post library preparation (diluted to 5µM), preferably with the annealing temperature used by the customer.
Paired-end 50 cycle sequencing is typically performed on the NovaSeq 6000 for ChIP-seq libraries (libraries can also be sequenced on a S4 PE-150 shared flow cell), with the following read recommendations:
Histone modification profiling:
- “narrow” peaks: at least 25 million reads/sample
- “broad” peaks: at least 50 million reads/sample
Transcription factor mapping: at least 25 million reads/sample
Questions?
Contact Us
1150 West Medical Center Drive
Ann Arbor, MI 48109