IRBMED Frequently Asked Questions (FAQ)

Answers to the IRBMED questions we hear most from faculty and staff.
IRBMED Fee
Beginning July 1, 2021, IRBMED’s application review fees are adjusting. The fee for IRBMED review of industry-sponsored studies will increase to $3750. Also on July 1, IRBMED administrative review of industry-sponsored studies ceded to a commercial IRB will increase to $1300. Contracts under active negotiation are expected to begin including these provisions to meet this deadline. Studies previously negotiated with contracts awarded after July 1, 2021, will be charged at prior rates. Of note: the new rates will also apply to all units across campus using IRBMED services and will no longer be limited to the Medical School.
For more information about the fees and local budgeting, please refer to our internally facing IRB website. Language to share with sponsors during the negotiation process is available in Section V: Fees and Budgeting for Industry-Sponsored Research on our externally facing IRB website.
IRBMED has also instituted approved review fees in association with single IRB (sIRB) reviews for multi-site research when IRBMED is the IRB of Record for the study. More information about the fee structure for qualifying studies and budgeting for these fees in grant applications and contracts is located in our externally facing website in Section II: IRBMED Accepts Oversight (sIRB) and Associated Fees.
For Questions:
- IRBMED review – Email IRBMED
- Contract negotiation – Clinical Trials Support Units or your local research administrator
- General sponsor applicability and reflecting IRBMED Fees/recovery in your sponsored project budget – Email Grant Services & Analysis Office
Why an IRB? Which IRB?
Please contact the IRB for assistance in determining whether your project requires review. The U-M HRPP Operations Manual Part 4 also provides information.
OHRP regulations and the Common Rule make IRBs responsible for review and approval of "human subjects research." This includes "clinical investigations" also regulated by FDA. Research is defined as "a systematic investigation, including research development, testing and evaluation, designed to develop or contribute to generalizable knowledge." A human subject is "a living individual about whom an investigator (whether professional or student) conducting research obtains (1) data through intervention or interaction with the individual, or (2) identifiable private information."
FDA regulations and policies also make IRBs responsible for oversight of some non-research activities, including Humanitarian Use Devices and Expanded Access (compassionate use).
IRBs enforce HIPAA regulations about the use of Private Health Information for research purposes.
Some activities that feature research methodologies are not regulated under OHRP or FDA regulations (e.g. case studies, analyses of publicly available datasets, research on organizations, quality assurance/improvement activities, standard public health surveillance, oral histories, journalism, etc.). Importantly, these projects may still be subject to HIPAA regulations.
You are required to submit an IRB application, and wait for IRB review and approval, before doing any “research” that also “involves human subjects.” Research is defined as "a systematic investigation, including research development, testing and evaluation, designed to develop or contribute to generalizable knowledge." A human subject is "a living individual about whom an investigator (whether professional or student) conducting research obtains (1) data through intervention or interaction with the individual, or (2) identifiable private information."
Some non-research activities also require IRB oversight; see “What kind of research…?” question above.
Some “human subjects research” is eligible for Exemption, which a determination of Exemption is required before the research starts, but there is no ongoing IRB oversight.
You are required to submit an IRB application, and wait for IRB review and approval, before doing any “research” that also “involves human subjects.” Journals require documentation of IRB approval for publications about human subjects studies. However, people seek to publish descriptions of nonresearch activities for a variety of reasons, if they believe others may be interested in learning about those activities. Quality improvement projects that were not undertaken for research purposes, as well as case studies, do not require IRB approval, and publishing the results do not require IRB review. See also FAQ on Not Regulated projects.
If your primary appointment is with the Health System, Medical School, Dental School, or School of Nursing or your research involves the health system patients or facilities, IRBMED would have jurisdiction over your application. IRB jurisdiction may also depend on the nature of the research and/or the expertise required to conduct a review.
The U-M HRPP Operations Manual Part 5.II also provides information on IRB jurisdiction.
For multi-site studies, a single “central IRB” may provide IRB oversight trial-wide: the central IRB is “IRB-of-record” for all sites. U-M has procedures for “ceding IRB oversight” from IRBMED to an external IRB in the following circumstances:
- The central IRB is a commercial/independent IRB which has a Master Agreement with U-M to provide oversight for multi-site, industry-sponsored clinical trials
- generally not for Phase I studies
- available with selected commercial IRBs
- The central IRB is based at another institution (generally another academic centers), if ceding is deemed appropriate upon consultation with UMOR and IRBMED (e.g. if required by NIH grant) and the Authorization Agreement Process is used
NOTE THAT “ceding” IRB review does not lessen U-M researchers’ obligations to other research administration units within the U-M HRPP:
- Approval of an eResearch Regulatory Management (eRRM) application before study activity is required, through the streamlined application type “Request Review by non-UM IRB”
- Application must be maintained in an approved state through “continuing review” and other submissions, as appropriate
- Review by non-IRB research review units (e.g. “ancillary committees” such as CRAO, Research Pharmacy (also known as IDS) or Conflict of Interest Review Committee) may be indicated
For more information, see HRPP and Research A-Z webpages.
The U-M HRPP Operations Manual Part 5.III and IV also provides information on IRB review for multi-site studies.
Many of the tips below are covered in more detail in other FAQ questions, or in other guidance on the IRBMED website or the HRPP website.
- Study Team Members need to have PEERRS training completed prior to approval of an application. You should complete a web-based educational module about doing “human subjects research” through https://research-compliance.umich.edu/peerrs-portal/
- Applications for IRB review are entered into a web-based “smartform” system, eResearch Regulatory Management (eRRM). IRBMED has a basic overview about using eRRM online. There’s also links to several other specific resources on this page.
The “smartform” interface means that, when you’re filling out an application, your early answers determine what questions you are asked later. The intent is that you are only asked relevant questions. To make this more likely to work well, it’s very important to fill out questions only on system-required pages. There’s some more information about this in the “General workflow” heading at the page linked above.
- It is critical that eResearch applications and all supporting documents are consistent throughout. See the eResearch Cross-reference table. IRB regulatory staff will require clarification for inconsistencies.
- Short educational presentations (.ppt + voiceover) are available on the U-MIC page on regulatory topics, some of which also specifically address eResearch concerns. A few you might find useful:
- eResearch: Before We Get Started
- IRB Board Tip: Protected Health Information (PHI)
- IRB Board Tip: Documentation of Informed Consent
- IRB Board Tip: The HIPAA Privacy Rule: Requirements and Waivers
- Principal Investigator Responsibilities per the HRPP Operations Manual and the Common Rule
- More IRBMED guidance webpages are found here.
- Student researchers: You should have a Faculty Advisor on your application if you will be the listed Principal Investigator (PI), or you could list yourself as a “Co-Investigator” and the faculty advisor as the PI. See HRPP Operations Manual Part 6.I.F for details.
- Children in research: there are specific additional regulations and policies about informed consent and assent for parents and children when children are research subjects. See more information here.
- PHI: For any study that involves accessing (hearing/viewing) PHI, HIPAA regulations about medical data apply – even if the PHI is not recorded. See more information here.
Study Team Qualifications
The Program for Education and Evaluation in Responsible Research and Scholarship (PEERRS) is a web-based instruction and certification program. Certification is obtained by passing a short quiz that covers required modules.
IRBMED recommends that all study team members complete PEERRS Human Subjects certification, or obtain waivers for equivalent certification (requestable through the PEERRS Portal), to demonstrate completion of human subjects protection training.
The U-M Human Research Protections Program requires PEERRS certification for all PIs, Co-Is, faculty advisors, and study coordinators who conduct research with human subjects.
Michigan Medicine Corporate Compliance Office requires (level-2 login required) PEERRS certification for all study team members who access MiChart for research purposes.
Data Office for Clinical and Translational Research requires PEERRS certification for all study team members who access DataDirect or EMERSE.
You may request a waiver for equivalent certification through the PEERRS Portal.
Students, trainees, etc. may be listed as Principal Investigator (PI) on an application if they also list an eligible Faculty Advisor, or students may be listed as a “Co-Investigator” and the faculty advisor as the PI. See UM HRPP Operations Manual Part 6.I for details.
See also IRB-HSBS main webpage, “References and Resources” heading:
- Guide for Student Investigators
- Tips for Student Researchers
- Tips for Faculty Advisors
Study team members without U-M affiliation (“external collaborators”) can be added to the eResearch application after obtaining a U-M “Friend account.” Like any other study team member, external collaborators are expected to accept their role on the study, fill out questions about outside financial interests, and to obtain PEERRS certification (or waiver) if their study role and/or duties indicate this. (See other FAQs What is PEERRS…? and “…conflict of interest disclosure?”
External collaborators who are “engaged in the conduct of human subjects research” in a “regularly regulated” study (approved by expedited or full board process, not exempt or “not regulated”) should either.
- Provide documentation of oversight for their activities by a local IRB/ethics board, or
- use the Authorization Agreement process to request IRBMED provide direct oversight for their activities
See also the FAQs at the Authorization Agreement process website.
Sharing study data and/or specimens with external collaborators often also requires Data Use Agreement (also known as Data Sharing Agreement) and/or Materials Transfer Agreement. Please see ORSP Unfunded Agreement webpage for more information. These agreements are processed separately from any IRB application.
Submitting Applications & Documents
Applications are managed through the eResearch Regulatory Management (eRRM) system. For further information, see
- IRBMED resources
- eRRM tips webpage
- ITS eRRM resources:
- FAQ webpage
- Step-by-step training documents
- “Show help” available within eResearch applications
- “Sandbox” (on-campus or VPN access): a copy of eResearch designed to orient new users to the system and allow for exploring options: applications in Sandbox are NOT submitted for real regulatory review).
IRBMED offers educational workshops on eResearch as well as on regulatory topics.
Human Subjects research involving interaction or intervention… – Most research studies, especially involving any primary collection of data and/or specimens (survey, interview, physical exam, blood draw, biopsy). A few of these are eligible for Exempt determination (See FAQ heading “Other application types”).
Secondary research uses… – Most studies that do not involve any interaction or intervention with subjects, but focus on analyzing data and/or biospecimens (with or without identifiers). These may receive a range of regulatory determinations (See IRBMED guidance “Elements and Uses of a Repository”).
- Note: the dedicated Repository (REP) application pathway is not intended for an analysis application (See IRBMED guidance(link is external) page “FAQ: Repository applications”)
Activities not regulated as human subjects research – choose only if the project does not meet the regulatory definition of "research" and/or does not meet the regulatory definition of "involving human subjects.” (See FAQ headings “Other application types…” and "Why an IRB? Which IRB?")
- Note: secondary use analysis is usually better captured under “Secondary research uses…” type, even if the analysis is eligible for “not regulated” determination. (See IRBMED guidance “Elements and Uses of a Repository”)
The other application types are more rare and specialized:
- Projects lacking immediate plans (aka "Umbrella") – most often associated with an NIH "Just-in-Time" request, or sometimes an NIH Genomic Data Sharing "Institutional Certification" request. See also U-MIC "Umbrella Projects."
- Single-patient Expanded Access... – there are separate "Drug or Biologic" and "Device" versions for this application. See IRBMED page "FDA Expanded Access Program..." and contact MICHR IND / IDE Investigator Assistance Program (MIAP) by emailing [email protected]
- Humanitarian Use Device – See IRBMED webpage "Humanitarian Use Device Requirements..." and U-MIC "Humanitarian Use Devices (HUDs)."
- Requesting Review by a Non-UM IRB (aka "Ceding") and Multi-site Research where U-M is a Coordinating Center and/or IRB of Record (aka "Single IRB or sIRB") – See
- IRBMED webpage Multi-site Research;
- U-MICs "Central Institutional Review Boards," "Collaborator Agreements," and "Single Institutional Review Board...," ; and
- U-M HRPP webpage Single IRB-of-Record Process
Exemption eligibility depends on the study using only research methodologies specifically designated as “Exemptable” in the OHRP regulations 45CFR46.104 or by U-M HRPP Flexibility Initiative policy. Many simple studies (e.g. one-time blood draws, or survey + medical record review) do not fit into any Exemption category/ies.
Generally, an IRB application for comprehensive review and approval requires more information from the study team than an application for Exemption, and is more likely to require Amendment during the study ‘lifecycle.’ However, some comprehensively reviewed and approved studies are eligible for “no continuing review required.”
Note: “Exempt” does not mean the same thing as “Expedited,” and does not mean the same thing as “not regulated.” See also FAQ headings “Other application types,” "Review & Approval Process," and "Ongoing Review."
IRBMED strongly encourages the use of IRBMED templates. Sample templates that contain all necessary regulatory information are available online. IRBMED offers a Standard Template that can be used for studies of all risk levels, as well as Specialty Templates for some simple, low-risk studies. Contact your regulatory staff to determine which consent template is most appropriate for your study.
Please see ITS guidance for uploading documents into eResearch
Please refer to the step-by-step guidance on 'stacking' documents. “Upload revision” button in eResearch puts a new document ‘stacked’ on top of previous version(s) while maintaining a full history of all versions uploaded. Documents previously uploaded to eResearch should be retained for historical and regulatory reference purposes, and should not be deleted.
For updating informed consent documents:
- Download from Section 10-1 of the eResearch application the most recent IRB-approved clean version of a consent
- Turn on Microsoft Word “Track Changes” functionality
- Make all necessary changes
- Upload the tracked-changes version into Section 10-1, maintaining the IRBMED standard naming conventions
Further instructions on updating informed consent documents are available at:
- Cover page in any IRB-approved clean version of a consent downloaded from Section 10-1 of the eResearch application
- IRBMED Statement of Practice “Version Control of Informed Consent Documents”
- U-MIC presentation on "Version Control of Informed Consent Documents"
- Instructions for Amendments at Standard Informed Consent Template guidance page
Application Content
Study teams should expect IRB staff to ask for confirmation when an eResearch application indicates data is “de-identified.” The term “de-identified” or “anonymized” should be used only to describe data that
- Previously contained identifiers, which have been stripped, and
- No “code” linking the data back to identifiers still exists, such that
- The dataset cannot be “re-identified” by anyone.
Data is not “de-identified,” but “coded” and “indirectly identifiable” if:
- Researchers have “coded” a data and store the code separately from the research dataset, or
- The data provider has the “key” to “coded” data, even if the researchers do not, or
- Anyone can “re-identify” the data by linking the dataset to direct identifiers.
U-M uses “de-identified” in this way to avoid confusion about whether data are “coded” or not, and in accordance with OHRP definitions and guidance. For further clarification, see the U-MIC educational presentation "Anonymous, Coded, and De-identified Data..." or the "Key Definitions" heading at the U-M HRPP Data Security Guidelines webpage.
Caveat: HIPAA Privacy Rule has a different definition of “de-identified,” applicable only to data based on Protected Health Information (usually medical records). Please specify “HIPAA-de-identified” if this definition is intended.
"Reimbursement" is compensating participants for specific out-of-pocket expenses, which should be documented by receipts the subjects provide to study team. If a set amount is provided to each participant for a given study visit, this is "payment."
For example, if each participant receives $20 this should not be characterized as “reimbursement for travel," since one participant's travel expenses could be a round-trip AAATA bus ride ($3), while another's could be the cost of gas for driving from and to Saginaw plus parking here (closer to $25).
See also HRPP guidance Payments to Research Participants.
Information about money provided to participants (when, how, how much) should be clear and consistent in the Informed Consent Document(s) (section 8 if using the IRBMED Standard Template) and in eResearch application section 13, and other sections as applicable. You may find the "eResearch Cross-reference table" available through Statements of Practice webpage of use.
The Health Insurance Portability and Accountability Act (HIPAA) Privacy Rule governs the use and release of a patient's personal health information, also known as Protected Health Information (PHI), by a covered entity. IRBMED studies most commonly involve access to PHI for at least some of the study activities, for instance:
- Medical record review and/or use of clinic schedule records to identify eligible subjects
- Data collection of clinical variables from the medical record (by direct chart review and also by tools such as EMERSE or DataDirect)
- Clinical trials providing study-related treatment at UMHS
- Receiving external PHI as part of multi-site data collection (often with a Data Use Agreement/Data Sharing Agreement)
HIPAA considerations are addressed mostly in eResearch Regulatory Management section 25 and sub-sections.
Data directly provided by a subject to a U-M researcher for research-only purposes is not considered Protected Health Information (PHI). This holds true even if the data provided by the subject concerns the health or medical history of the subject or a family member. As such, surveys for research purposes usually do not create PHI. However, any identifiable information that is added to the medical record is governed by HIPAA.
Survey-based studies may access PHI in the course of a study for several reasons, for instance:
- Medical record review to identify eligible subjects for recruitment purposes (usually under a Waiver of HIPAA Authorization)
- Survey responses are correlated with individual Michigan Medicine health data, for instance to explore patient recall of certain care conversations (usually with individual signed HIPAA authorization, often incorporated in the IRBMED Specialty Informed Consent Template for Survey Research)
- “Patient-reported outcome” measures are used as part of a clinical trial and the results are entered into the medical record (usually with individual signed HIPAA authorization incorporated into the IRBMED Standard Informed Consent Template)
IRBMED studies most commonly involve access to PHI for at least some of the study activities.
MyUofMHealth.org (also known as Patient Portal) is designed for Michigan Medicine patients to access their medical records, and for communication between patients and providers/clinics.
For enrolled participants (patient has an 'Active' enrollment status in MiChart), the study team can request permission from participants to communicate through Portal:
- For studies that require Portal participation by participants as a condition of being in the study (e.g. communications through the portal are not duplicated in other formats), prospective permission should be sought explicitly in the Informed Consent Document(s).
- For studies that allow for Portal use for convenience, but for which use of the Portal is not a required condition of study participation, the study team can seek permission in a less formal correspondence or conversation, and document permission in research files.
- Communication through the portal is allowed to subjects who have consented to this communication method as part of a specific study (e.g. sending messages about upcoming research appointments, sharing new versions of informed consent documents prior to research visits, etc.). The Portal automatically sends a generic “You have new information in MyUofMHealth” notification; all specifics of the message are available only upon participant login to the Portal. As such, this is not an appropriate method for time-sensitive or critical communications.
- For some Michigan Medicine studies, participants (with an 'Active' enrollment status in MiChart) can access basic study information (title from eResearch, PI and study coordinators) in MyUofMHealth.org. Further information about this functionality is at Michart Research Resources (level-2 login required), “Research Study Visibility” heading).
For potential participants, Send Recruitment Requests to the Portal via MiChart:
- Researchers (study teams) may request that the MiChart Research team activate the Patient Portal workflow: Research - Use of the MiChart Patient Portal for Research Recruitment (level-2 login required). Patients should not be contacted in regards to recruitment using any other Patient Portal method.
As of May 2026, the IRB eResearch application includes new questions to capture the use of AI technology in studies involving human participants. IRB staff and reviewers will evaluate the appropriateness of AI use in accordance with University guidance and standard review considerations.
When AI is used in the conduct of human research, study teams should clearly describe its role in the eResearch application, protocol, and consent materials. This includes outlining the AI’s function, the data it uses and generates, how outputs will be applied, and the safeguards and human oversight in place.
Additional guidance is available on the HRPP AI guidance webpage, including examples of AI use, considerations for protocols and consent documents, third-party tool guidance, amendment triggers, and links to U‑M ITS Safe Computing resources for AI and sensitive data.
A subset of AI-enabled Device Software Functions qualify as FDA-regulated medical devices. Contact your IRB staff or MICHR-MIAP with questions about FDA regulation of device software functions.
Review & Approval Process
According to federal regulations, studies that pose greater than minimal risk to subjects must be reviewed at a Convened Board meeting. Sensitive or complex studies, as well as those involving vulnerable populations, might also require full board review.
Generally you should be able to expect a decision within 4-8 weeks for full board review, and 2-4 weeks for expedited review. Please alert the IRB if your application is time-sensitive.
IRBMED staff makes every effort to complete initial processing on an application within 2 weeks of receipt to IRBMED. Total time to approval will vary with an individual application's review needs, including additional reviews, application completeness and clarity at initial submission, and IRB Board reviewer schedules.
PLEASE NOTE: some applications, particularly clinical trials, require eResearch review and approval from non-IRB U-M research review units (sometimes called "Ancillary units") after submission by the study team and before receipt by IRBMED.
IRBMED processes submissions on a ‘first-in, first-out’ basis. However, if there is a particular time pressure about a submission, please alert your IRB regulatory staff.
Please note that "expedited review" means review by one IRB member on behalf of the Board. Expedited review is available for some human subjects research applications, based on risk level and certain other characteristics. It does NOT necessarily imply faster processing.
Determination notices are issued via eResearch. You will receive an email notification with a link to your approved study in eResearch. You also can track a submitted application through its eResearch progress; the step-by-step guide Tracking a Submitted Application shows how.
No. You may begin only after the IRB has determined that all contingencies have been resolved (as evidenced by an “Approval” determination notice). The step-by-step guide Study Approval and Contingencies provides information on making requested changes to an application "approved with contingencies." Your IRB regulatory staff can also help.
The current IRB determination letter (“Approved,” “Exempt” or “Not Regulated”) is available under the “Activities and Correspondence” heading of
- the main study space (aka HUM space) before an Amendment or Continuing Review is finalized
- the submission workspace for the most recent completed Amendment (Ame) or Continuing Review (CR)
The HUM workspace shows dates for the Last CR and Last Amendment approval dates. If both are blank, the letter in the main HUM workspace is current. The study in the example below has had a CR but no Amendments (“Last Amendment” is blank).
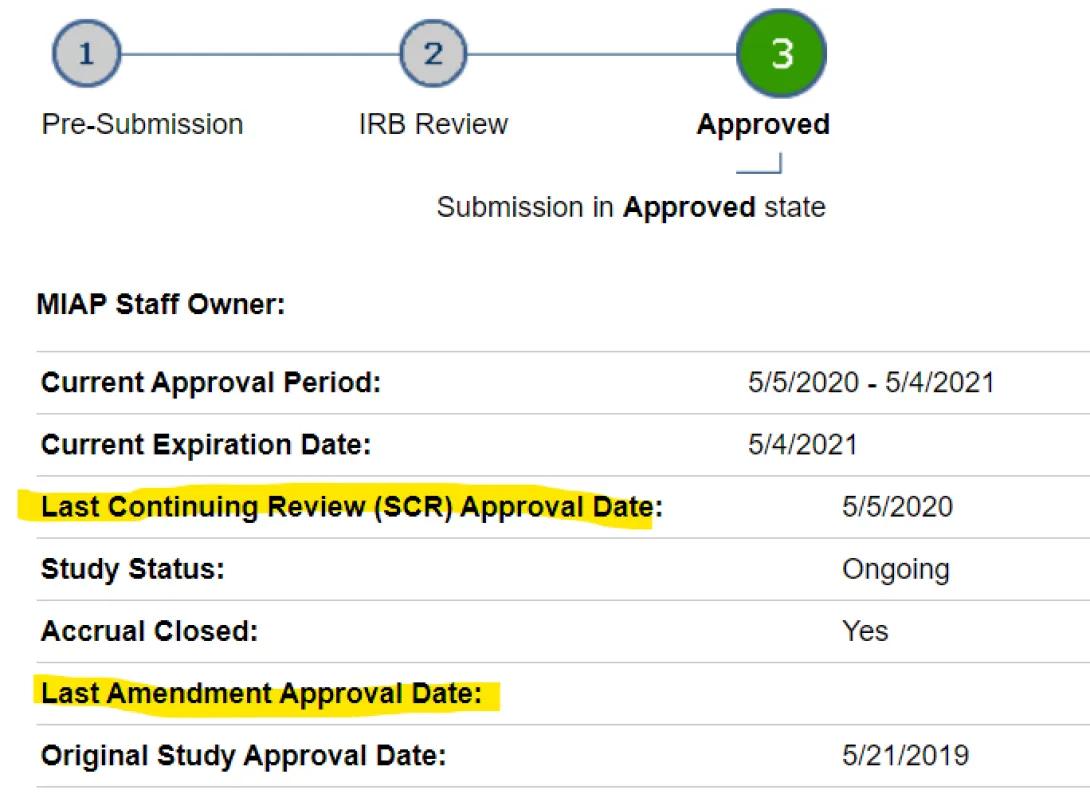
Go to the deep blue tab near the top of the HUM workspace for "Amendments" or “Continuing Reviews,” whichever is most recent. Click into the individual submission workspace, scroll down to Activities and Correspondence heading, and find the "See Approval Letter" link (or “Exempt” or “Not Regulated”). The letter opens in a new tab, from which you can print or "print-to-.pdf" a copy.
Contact the IRBMED regulatory staff assigned to your application if something appears to be missing or mistaken on a determination notice you have received.
Sponsors may require specific language in determination notices, such as the version number and date for the Protocol reviewed by the IRB. In these cases, study teams should enter the desired language in eResearch field 44.2 (new Application or Amendment) or 15.1 (AE or ORIO report). This text prints word-for-word on the determination notice as "Supporting Documents." To include specific language in a Continuing Review determination notice, contact the IRBMED regulatory staff assigned to your application.
eResearch determination notices list the ID and may list the expiration date for the U-M FWA. It is not a regulatory deficiency for you to present to a sponsor or journal a notice showing an apparently lapsed FWA, as long as the date shown was current at notice issuance. For the current U-M FWA expiration date, use the OHRP web search: enter FWA Number: 00004969 or Institution/Organization Name: U Michigan.
Ongoing Review
Any change to an approved, non-exempt study requires submission of an amendment application via eResearch. Examples of such changes include, but are not limited to:
- alteration of study design, methodology, or recruitment methods
- changes to surveys
- changes to consent documents
- addition/deletion of principal investigators or key personnel
- addition/deletion of research performance sites
- alteration of project title
Some approved, non-exempt studies that do not require continuing review (see also FAQ "How long is my approval valid?"): amendments are still required for these studies.
Exempt studies and “not regulated” projects do not require amendment unless the proposed change exceeds the scope of the determined category/ies, or unless ancillary review is indicated.
See also FAQ “How do I update a previously uploaded document?”
Go to your approved application within eResearch and click “New Amendment” under “Create New Submission.” An Amendment must include both a "Cover Sheet" and changes to the eResearch application. The step-by-step guide Creating an Amendment provides useful information on the Amendment process.
If an approved study space shows an Expiration Date, the study requires "continuing review" and re-approval at regular intervals. The approval period is usually one year. Approval can only be extended by IRB approval of a “continuing review” application (see FAQ “How do I apply for a Scheduled Continuing Review?” and "What happens if my approval expires...?").
Some ongoing studies do not require annual Continuing Review (CR) per Revised Common Rule or per Flexibility Initiative. The study workspace indicates “No Continuing Review Required for this application” when applicable.
eResearch sends notifications prior to expiration of study approval (at 90, 60, and 30 day intervals). To submit a Scheduled Continuing Review (SCR), go to your application within eResearch and click “New Continuing Review” under “Create New Submission.” The step-by-step guide Creating a Continuing Review also provides information on this submission type. The IRB will review your application and notify you of the outcome via eResearch.
Yes; in eResearch these two types of submissions can be processed concurrently. Only one Amendment and only one SCR active at any given time.
Per U-M IRB Standard Operating Procedures Part 3.III.C.4.b.3.a,
If the IRB has not reviewed and approved the continuing review application by the expiration date of the current approval (regardless of the reason or circumstances), the study will be considered lapsed.
- The research must stop unless the IRB finds it is in the best interest of individual subjects currently participating in the study to continue the research interventions or interactions.
- Enrollment of new subjects during a lapse is prohibited
- IRB will remind investigators that resources must not be expended for unallowable activities.
Lapses of IRB approval on a single study, or a pattern of lapses across multiple studies raise serious concerns. Non-compliance with federal or institutional requirements to maintain continuous IRB approval for human subjects research may require corrective action including: review by the full IRB, additional education of the PI and study team, study monitoring, suspension of IRB approval status, and/or referral to higher institutional authorities or federal agencies. Suspension or termination of IRB approval is reportable to OVPR (Office of the Vice President for Research) and ORSP (Office of Research & Sponsored Projects) and may be reported to external regulatory agencies under federal regulations.
Submit a termination report via eResearch when
- research activity is limited to analysis of de-identified data (i.e., all identifiers, links, or codes have been destroyed), OR
- the research has ended.
Note that ‘de-identified’ means the dataset cannot be “re-identified” by anyone (See FAQ What does ‘de-identified’ mean?...).
Industry-sponsored studies: upload the study closure visit confirmation into the termination report.
Identifiability and confidentiality of study data/biospecimens: IRB responsibilities include determining that a study adequately protects the confidentiality of participants’ data; moreover, data confidentiality and data sharing is increasingly a concern among the public and sponsors. Hence, the Termination report prompts a review of the study’s previously approved plans for disposition of participant identifiers and of data/biospecimens after Termination. It is usually preferable to destroy links to identifiers as soon as practicable, and to retain study data for a period based on institutional policies, federal/state regulations, and/or sponsor obligations.
See ITS step-by-step guides for Termination of IRB-approved studies, and for exempt studies.
Contact UMMS Regulatory Affairs Office [email protected] for comprehensive guidance. The “Faculty Offboarding Checklist” linked from the Regulatory Affairs Resources for Researchers webpage (level-2 login required) is an excellent resource for departing faculty and their support people.
Active studies where the departing faculty is on the study team should be amended to change or remove his/her participation. Your IRB regulatory staff can help compile a full list of these studies. See step-by-step eResearch instructions Changing the PI on an application.
When the departing faculty will remain involved with the study (even if only analyzing “coded” or “de-identified” study data), new agreements may be necessary. Units supporting these agreements may include UMHS Compliance Office (level-2 login required), ORSP (particularly regarding research equipment or Data Use Agreements), and Innovation Partnerships (regarding Materials Transfer Agreements).
Other Required Reporting
Yes, follow the Standard Timetable unless
- a study-specific reporting plan has been approved, or
- the study is limited to retrospective data/specimen analysis without human subjects interaction.
Include a study-specific reporting plan in eResearch section 32-1 as part of a Data Safety and Monitoring Plan (DSMP). Until you receive a 'Notice of Outcome' indicating IRB approval of the study-specific plan, follow the Standard Timetable.
Study-specific AE reporting plans are often appropriate for
- Sponsored studies that include a detailed AE reporting plan for submitting adverse events to a sponsor and/or a data safety committee
- Minimal to moderate risk studies that do not fall under FDA oversight.
An ORIO is a way to tell the IRB about information or occurrences, other than adverse events, related to the conduct of your research. IRBMED requires reporting of some kinds of ORIOs. Study-specific ORIO reporting plans may also be appropriate for some studies, similar to study-specific AE reporting.
Additional Research Oversight
All Michigan Medicine Faculty, House Officers, and Staff must disclose outside interests or activity, including any potential Conflicts of Interest (COIs) and any potential Conflicts of Commitment (COCs) through MInform. The COI Committees reviews disclosures and, when necessary, works with the faculty or staff member to eliminate, minimize, or manage the conflict. An interface between MInform and eResearch Regulatory Management identifies study team members with possible COI.
Payment for research is taxable income. University Treasury office tracks payments to subjects and issues IRS 1099 forms if >$600 received by an individual in a calendar year. To comply with this, study teams should have subject payments processed through Human Subjects Incentive Program (HSIP). See http://spg.umich.edu/policy/501.07 HSIP should be involved in "regularly regulated," Exempt and Not Regulated studies unless they are using a third-party vendor, or another institution is handling the incentives.
See also HRPP guidance Payments to Research Participants.
There is a state requirement that many privately administered lotteries/raffles be licensed by the state. Please see HRPP guidance Payments to Research Participants and U-MIC presentation "Lotteries".
Human subjects research, especially clinical trials, often requires permissions and/or agreements beyond IRB approval. Although some types of additional agreements are referred to in the IRB application, they are not processed by IRB staff, nor finalized through IRB approval.
Faculty, chairs, and departmental administrators do not have authority to sign legal agreements on behalf of the University.
At University of Michigan,
- Unfunded Agreement (UFA) system processes Data Use Agreements (DUA, also known as Data Sharing Agreements or DSA), Business Associate Agreements (BAA), Materials Transfer Agreements (MTA), and some other contracts/agreements not directly related to funding
- Proposal Approval Form (PAF) system processes grants, contracts, and cooperative agreements for funding.
- Certificates of Confidentiality (from NIH or CDC) are processed through HRPP staff.
- Authorization Agreements for external collaborators in U-M research are processed through HRPP staff.
UMMS Regulatory Affairs maintains documentation regarding FDA Part 11 compliance at Data Integrity and Sharing (level-2 login required).
Before seeking approval from IRBMED, your work must comply with Information Assurance (IA) guidelines. Adhering to security protocols is mandatory when accessing Michigan Medicine's network. If the applications and technologies are not vetted for compliance beforehand, it may hinder the progression of your research project. Oversights have led to research schedule setbacks and, in some cases, could jeopardize the entire study if the systems, services, or vendors fail to meet Michigan Medicine's stringent security requirements.
Two important points:
- All technologies, such as research platforms and applications, that intend to connect to Michigan Medicine’s IT infrastructure need to be subjected to a review by Information Assurance (IA) and obtain clearance. This mandatory evaluation is necessary regardless of the types of research data, records, and patient information the technology utilizes.
- A Technical Owner must be designated for the IT system/service before IA can begin its security review
Researchers should identify a Technical Owner* early in their research design – ideally before IRBMED submission. A Technical Owner is typically an IT support person responsible for looking after technology and making sure it stays secure and compliant with Michigan Medicine IT security standards.
A Technical Owner’s first job is to contact the IA Department by filling out a request form called the Michigan Medicine Information Assurance Request (MMIAR) - Consult. Obtaining MMIAR approval is central to Michigan Medicine's vetting process for new technologies that could possibly introduce vulnerabilities into the environment. Early collaboration between research teams, Technical Owners, and the IA department ensures all research investments are supported, secure, and positioned to meet intended timelines.
*Most often, research studies are supported by the Academic IT Division within Health Information Technology and Services (HITS); however, resourcing and existing responsibilities may affect their ability to assign a Technical Owner to a research project at any given time. In some cases, an alternate Technical Owner may be arranged, but often a research study must wait until an IT support department has the resources available to assist.
Other Application Types
Exempt studies are a subset of human subjects research that do not require comprehensive and ongoing IRB oversight. Exempt status does not lessen the ethical obligations of researchers to subjects. For further information, see IRBMED Exempt Human Subjects Research webpage.
To be "not regulated," a project must NOT meet the regulatory definition of "research" at 45 CFR 46.102 (l) (Common Rule), and/or NOT meet the regulatory definition of "involving human subjects" at 45 CFR 46.102(f) (Common Rule) or at 21 CFR 56.102 (e) (FDA). See FAQ "Why an IRB? Which IRB?" heading for more information on these definitions. This means that some research involving medical data or specimens, and some other activities conducted by medical researchers, are "Activities Not Regulated as Human Subjects Research."
There is no OHRP, University of Michigan or IRBMED requirement for researchers to obtain a formal determination of "Not Regulated" (see U-M HRPP Operations Manual Part 4.V.A). However, if your project will be accessing/viewing/obtaining Private Health Information (PHI) for research purposes (analyzing a HIPAA Limited Data set, analyzing PHI from Decedents, or review of PHI preparatory to research), submit a "Not Regulated" application to ensure HIPAA compliance.
There is a shortened eResearch application path for this, beginning on page 01-1 "Application Type." Study teams may seek a "Not Regulated" determination through eResearch at any time; this is most common when you are uncertain whether the proposed activities should be regulated, or your funding requires input from the IRB.
Some “Not Regulated” categories permit a “self-determination” through eResearch, where you enter information into an application in eResearch, but the IRB never receives or reviews the application.
NOTE THAT a “not regulated project” is not the same as an “Exempt study,” although some of the requirements (and lack thereof) are similar.
Case study applications are limited to reports about ONE or TWO individuals. Because these cannot be assumed to be generalizable, case study reports are not regulated as "research." See U-M HRPP Operations Manual Part 4 (section I on "research" definition, section V on "not regulated" projects). The question of “research consent” as you have probably heard about it before regarding IRB oversight does not apply.
The use of medical charts (including protected health information or PHI) to write up case studies is regulated under the HIPAA Privacy Rule. It is common for journals to require HIPAA authorization (written permission) from patients whose cases are described in submitted articles. However, IRB regulations on Informed Consent (process and documentation) do not apply to case studies.
If it is feasible to obtain written HIPAA authorization from the patient, this is BY FAR PREFERABLE. Use the Michigan Medicine Permission to Release Information for Case Study Publication (level-2 login required). NOTE THAT a copy of the signed authorization should be sent to the Health Information Management (HIM) Release of Information Unit (3621 S. State Street, 700 KMS Place, Ann Arbor, MI 48108-1633, fax 734-936-8571) for processing and inclusion in the patient's medical record per Michigan Medicine Policy 01-04-310 (level-2 login required).
HIPAA authorization is not required (though still PREFERABLE) if you will REMOVE all HIPAA identifiers ("de-identify") from the write-up or presentation of the case, AND if you have no reason to believe the case is unique enough to be identifiable by the characteristics described.
Consult also Research A-Z webpage Quality Assurance and Quality Improvement (QA/QI) Projects.
IRB approval is usually not needed to perform quality improvement/quality assurance (QI/QA) activities or to present or publish QI results internally or externally, because QI/QA does not usually meet the regulatory definition of "research" at 45 CFR 46.102 (l) (Common Rule). However, OHRP FAQ on QI define “QI activities” more narrowly than the term QI is sometimes used. You should understand those technical definitions and be sure your project does not include sufficient “research” components that IRB approval is needed before you initiate the project. NOTE WELL that projects undertaken jointly with QA/QI and research intent (“research on QI” or “research with QI”) DO require regulation as research. Examples include the CDC Prevention Epicenters Program and Dr. Peter Pronovost’s Indwelling Catheter QI Procedures.
If you wish to obtain IRB input on whether the regulations apply to your QA/QI project, fill out an abbreviated eResearch application describing the project (“Activities Not Regulated…” application type), and submit it to IRBMED. IRB staff are also available by phone or email for consultation: ask for the Exempt/Not Regulated Coordinator. An informative overview is available from UMMS Maintenance of Certification (MOC) Program on presenting and/or publishing QI (Section 6 of that page is about IRB considerations).
IRB strongly recommends you submit an application to confirm “Not Regulated” determination for QA/QI activities if these involve data analysis from multiple sites, or through a registry of clinical data (e.g. a national health registry), or if the QA/QI activities involve multiple methodologies (e.g. data collection from medical charts and educational intervention(s) for providers).
A "system-generated determination" (also known as "Self-determination") option appears at the "Section 45 End of application" screen for some Pre-Submission HUM applications requesting Exemption or Not Regulated determination. The eResearch system offers the "system-generated determination" option only if the information provided indicates that
- the eligibility for Exemption (or "not regulated" determination), and
- the inapplicability of additional regulatory requirements such as HIPAA or FERPA,
are so settled that personal review of the application by IRB staff would not substantively add to research participant protection.
The PI has the choice to "Submit to IRB" rather than take advantage of "System-generate Determination Letter" for formal confirmation that the form questions are answered accurately. After an application is Submitted to IRB, the "system-generate determination" option is no longer available. A PI who wishes formal input from the IRB after a "system-generated determination" can Create an Amendment and "Submit to IRB."
The "System-generate exemption" process is described at
- IRBMED guidance webpage "Exempt human subjects research" (heading “What is an Exemption,” subheading "How are Exempt studies regulated?")
- U-M HRPP Operations Manual Part 4.VI.C
- for an eligible Pre-submission HUM application, at the "Section 45 End of application" screen, and
- for a finalized "System-generated Exemption," at the "View ... Letter" link in the HUM workspace "Activities and Correspondence" heading
Mockups of the smartform pages show how responses affect eligibility for system-generated exemption:
For Exempt studies Termination comprises a simple single pop-up screen, and can be completed by any study team member with edit rights. See ITS step-by-step guide "Creating a Termination - Submit a request for terminating an exempt study." Exempt studies receive an "Annual Touch Point" reminder of the option to Terminate on the anniversary of the Exempt determination.
Not Regulated projects need not and cannot be formally closed in the eResearch system.
Exempt Termination is not a regulatory requirement, but an opportunity to improve the HRPP's 'picture' of ongoing research.
Repositories
For information on IRB oversight of data and/or biospecimen repositories, see website guidance Repository Overview and sub-pages, including FAQ: Repository Application (REP).
Contact Us
University of Michigan Medical School
Ann Arbor, Michigan
About Us

We're Accredited
IRBMED has full accreditation by they Association for the Accreditation of Human Research Protection Programs, Inc.